


La enfermedad de Pyle (OMIN número 265900) es una displasia metafisaria de curso benigno que se hereda con un patrón autosómico recesivo. Se han descrito unos 30 casos genuinos hasta el momento. La causa de este proceso se conoce desde 2016, cuando se descubre su relación con mutaciones en el gen que codifica la proteína sFRP, un conocido inhibidor de la vía Wnt. Se presenta el caso de un varón de 58 años, diagnosticado de enfermedad de Pyle con base en sus características clínicas y radiográficas, cuyo fenotipo muestra un control diferencial de la homeostasis del hueso cortical y trabecular.
Pyle's disease (OMIN number 265900) is a metaphyseal dysplasia of benign course, inherited with an autosomal recessive pattern. Some 30 genuine cases have been described so far. The cause of this process has been known since 2016, when its relationship to mutations in the gene encoding the sFRP protein, a known inhibitor of the Wnt pathway, was discovered. We report the case of a 58-year-old man, diagnosed with Pyle's disease based on his clinical and radiographic characteristics, whose phenotype suggested a differential control of cortical and trabecular bone homeostasis.
La enfermedad de Pyle (EP, OMIN número 265900) es una displasia metafisaria de curso benigno1 que se hereda con un patrón autosómico recesivo2. Fue descrita en 1931 por Edwin Pyle3, cirujano ortopédico de Connecticut (EE. UU.), en un varón de 5 años que presentaba genu valgo severo en ambas rodillas y deformidades craneales. Desde entonces se han descrito unos 30 casos genuinos, en diferentes países y continentes. En la revisión efectuada en las bases de datos PubMed y Embase solamente hemos encontrado un caso publicado en España4.
A pesar de su rareza, el interés clínico por la EP ha aumentado recientemente al descubrirse su relación con mutaciones en un inhibidor soluble de la vía Wnt5–7. El análisis del fenotipo de estos pacientes y los hallazgos observados en modelos murinos que reproducen la disfunción del gen sFRP evidencian un control diferencial en la homeostasis del hueso cortical y trabecular. A continuación se presenta un caso con imágenes características y se comenta el relevante interés de las manifestaciones clínicas de estos pacientes.
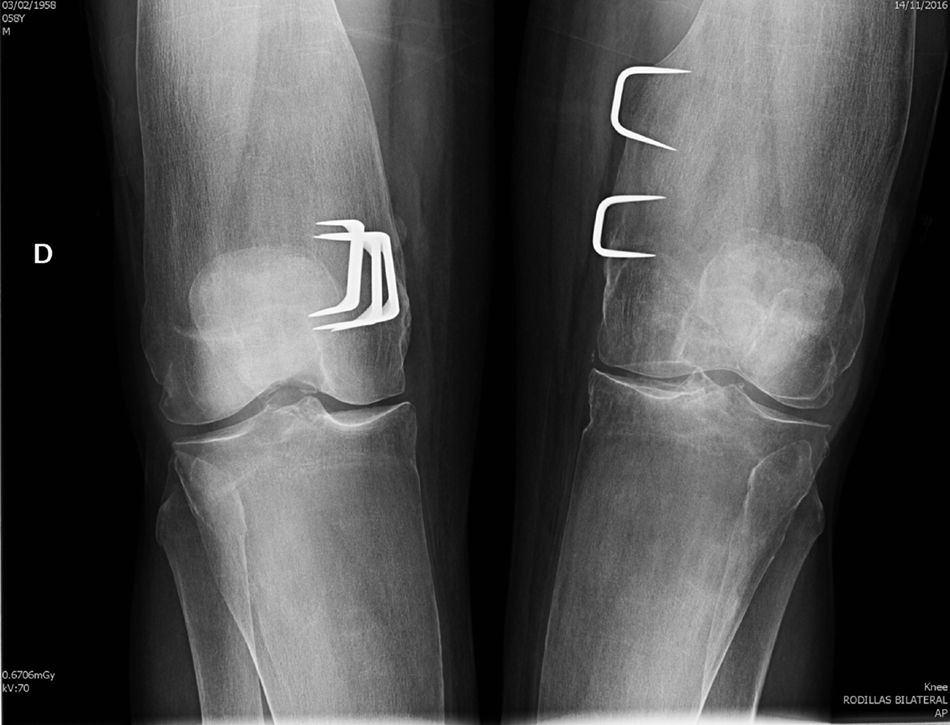
Observación clínicaSe trata de un varón de 58 años, enviado a la consulta externa de Reumatología por sospecha de fragilidad ósea. A los 6 años de edad había sido intervenido de genu valgo bilateral marcado, sospechándose entonces una osteopatía congénita. La intervención mejoró la estática de sus miembros inferiores y el paciente pudo realizar una vida normal, sin procesos patológicos relevantes, hasta los 57 años, cuando, tras un traumatismo de baja intensidad, sufre una fractura de clavícula y cotilo izquierdos. En la anamnesis efectuada no se recogieron factores de riesgo de osteoporosis ni antecedentes familiares o personales de interés, excepto la intervención quirúrgica comentada. En la exploración física se trataba de un adulto con buen aspecto general, sin deformidades craneofaciales, con un moderado genu valgo bilateral y las limitaciones propias de las fracturas recientes, para las que precisó tratamiento quirúrgico. Presentaba una talla de 182cm, con 81kg de peso (IMC 24,45kg/m2), resultando el resto de la exploración física irrelevante. Los análisis generales resultaron normales, con cifras de calcio iónico sérico de 2,41mmol/l, fosfatasa alcalina total de 115u/l, 25-OH-D3 de 32,8ng/ml y PTHi de 56pg/ml. La densitometría ósea mostró un T-score de −1,4DE en columna lumbar (928mg/cm2) y de −2,2DE en cuello femoral (625mg/cm2). En el estudio radiográfico realizado se objetivó un patrón característico de la EP, con un ensanchamiento metafisario de los huesos tubulares (fig. 1), un marcado adelgazamiento cortical y normalidad en la morfología epifisaria.
Discusión
Nuestro paciente cumple las características radiográficas de la EP en el adulto, que incluyen una expansión metafisaria de los huesos largos y un marcado adelgazamiento cortical8,9. Todos los huesos largos pueden estar afectados, aunque los hallazgos característicos son más evidentes en la tibia proximal y en el fémur distal (con el típico aspecto en «matraz de Erlenmeyer»). Los huesos craneofaciales pueden verse afectados con cambios discretos y ocasionales, como osteoesclerosis parcheada y obliteración de los senos paranasales. No obstante, cuando estas alteraciones son marcadas deben descartarse otras enfermedades hereditarias, sobre todo la displasia craneometafisaria, que comparte con la EP el modelado anormal metafisario, pero se diferencia claramente ya que los pacientes afectados presentan una facies muy peculiar, con hipertelorismo y engrosamiento del puente nasal, y no tienen afectación de clavículas, costillas y ramas isquiopubianas. La enfermedad de Camurati-Engelmann también se incluye en el grupo de las displasias metafisarias, pero, a diferencia de la EP, provoca hiperostosis en la base del cráneo y engrosamiento cortical en los huesos largos. Otros procesos a considerar en el diagnóstico diferencial de la EP son las osteopetrosis, en las que, además del aumento de densidad ósea que caracteriza al grupo, se aprecian imágenes de «hueso dentro de hueso» tanto en los cuerpos vertebrales como en las regiones diafisometafisarias de los huesos tubulares. Otras enfermedades a tener en cuenta en el diagnóstico diferencial son la enfermedad de Gaucher, la de Niemann-Pick y las talasemias, aunque son fácilmente descartadas por la historia clínica y las pruebas complementarias, así como por la ausencia de afectación extraósea y la normalidad de las pruebas de laboratorio que se observan en la EP.
Las fracturas por fragilidad son frecuentes en la EP y se localizan en las metáfisis con corticales adelgazadas, con especial preferencia por los lugares de transición entre el hueso displásico metafisario y el hueso diafisario, como se pudo observar en la fractura de clavícula de nuestro paciente. En la EP está afectada la arquitectura cortical, pero el hueso trabecular es normal, y su densidad puede, incluso, estar aumentada. Este sorprendente perfil fenotípico induce a especular con una homeostasis diferenciada entre el compartimento cortical y el trabecular, cuyo origen molecular ha permanecido oculto hasta el momento y que revisamos brevemente a continuación.
En 2016, Simsek Kiper et al.5 realizaron una secuenciación del exoma completo de 4 pacientes con EP, identificando una mutación «nonsense» bialélica en el gen sFRP4 (606570.0001) que codifica la proteína relacionada con Frizzled segregada 4 (sFRP4), un conocido inhibidor de la vía Wnt. Este defecto fue confirmado por otros 2 grupos de forma casi simultánea6,7. En un modelo murino de comprobación5 se anuló el gen sFRP4, obteniéndose una progenie con corticales muy adelgazadas tanto en los huesos largos como en los planos desde una edad muy temprana, y un aumento del volumen trabecular y de los parámetros dinámicos de formación trabecular, con una reducción de la tasa de aposición endocortical y subperióstica. El número de osteoclastos era normal en el hueso trabecular, pero estaba aumentado en la superficie endocortical diafisaria. Todos estos hallazgos son característicos de la EP humana y evidencian que la disfunción de sFRP4 provoca un hueso trabecular aumentado (como ocurre en otras enfermedades con liberación de la vía Wnt10), pero, de manera imprevista, un grosor cortical reducido y un fallo del modelado cortical durante el crecimiento, que conduce a un ensanchamiento metafisario y una fragilidad ósea que se mantiene en el adulto.
La vía Wnt es el principal sistema de señales que controla la renovación tisular11,12. Sus miembros actúan a nivel local, mediante señales específicas de tejido, dirigiendo el destino de las células progenitoras. Está fuertemente regulada por un amplio número de efectores, que se comportan como agonistas o antagonistas que actúan a nivel intracelular, modulando la transducción de la señal, o a nivel extracelular, regulando las interacciones ligando-receptor13. La familia de proteínas sFRP es la más numerosa entre los inhibidores segregados de la señal Wnt extracelular. En humanos se conocen 5 miembros (sFRP1-5). Todos ellos comparten un dominio de estructura similar al cystein-rich domain de unión a Wnt de los receptores Frizzled, que facilita el secuestro de los ligandos Wnt, impidiendo su unión al receptor. Existen pruebas de un modelo de inhibición alternativo, mediante el cual las sFRP formarían complejos no funcionales con estos receptores, impidiendo su unión al ligando Wnt14. A través de ambos sistemas, las sFRP pueden inhibir tanto la vía canónica como la no canónica de activación de la señal Wnt, participando de manera muy relevante en el modelado y remodelado óseos en la etapa posnatal, cuya disfunción alcanza su máxima expresión clínica en la EP15. En este proceso, el hueso trabecular presenta un fenotipo similar a otros que se caracterizan por un incremento de la señal Wnt canónica. Sin embargo, el hueso cortical muestra un marcado adelgazamiento, ya presente desde las primeras etapas de la maduración esquelética, un hallazgo contrario a lo esperado cuando liberamos la señal Wnt de uno de sus principales inhibidores y que evidencia que el hueso trabecular y el cortical tienen mecanismos de regulación y control homeostático diferenciales.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


