


Las enfermedades autoinflamatorias sistémicas engloban un conjunto de enfermedades poco frecuentes caracterizadas todas ellas por la presencia de episodios inflamatorios agudos y recurrentes, que son consecuencia de una disregulación del control del proceso inflamatorio. Desde sus respectivas descripciones clínicas, se había observado un claro patrón hereditario mendeliano para algunas de ellas. En fechas recientes se han identificado los defectos genéticos y moleculares subyacentes al identificarse mutaciones responsables de enfermedad en diferentes genes relacionados con la respuesta inmune innata y con la inflamación. A lo largo de la presente revisión se abordarán de una manera actualizada los principales aspectos clínicos, fisiopatológicos y terapéuticos de las diferentes enfermedades autoinflamatorias hereditarias.
Systemic autoinflammatory diseases encompass different rare clinical entities characterized by recurrent acute inflammatory episodes secondary to a dysregulated inflammatory process. Since their first clinical descriptions, the Mendelian hereditary nature of some of them became evident, with their genetic and molecular basis being recently elucidated. There are disease-causing mutations in genes encoding for different proteins involved in the innate immune response and inflammation. Herein, we will introduce the reader to an updated review of the main clinical, physiopathological and therapeutic features of the different hereditary systemic autoinflammatory diseases.
El concepto de enfermedad autoinflamatoria sistémica fue propuesto en 1999 por el Dr. Kastner, del National Institute of Arthritis and Muskuloeskeletal and Skin Diseases (NIAMS), para agrupar unas enfermedades con manifestaciones clínicas y bases fisiopatológicas aparentemente similares (episodios febriles e inflamatorios recurrentes). Desde su aparición, se estableció una contraposición entre dichas enfermedades y las enfermedades autoinmunes, con las cuales pueden compartir ciertas semejanzas clínicas, pero con las que presentan evidentes diferencias fisiopatológicas, al no detectarse en las enfermedades autoinflamatorias los marcadores de una respuesta autoinmune (autoanticuerpos a títulos elevados o células T específicas de antígenos propios)1. En la actualidad, se propone la disregulación del proceso inflamatorio como base fisiopatológica común para todas las enfermedades autoinflamatorias. Como veremos a lo largo de esta revisión, algunas de estas enfermedades autoinflamatorias son hereditarias, con un típico patrón mendeliano, consecuencia de mutaciones que afectan a genes codificantes para proteínas implicadas directamente con la inflamación y su regulación2.
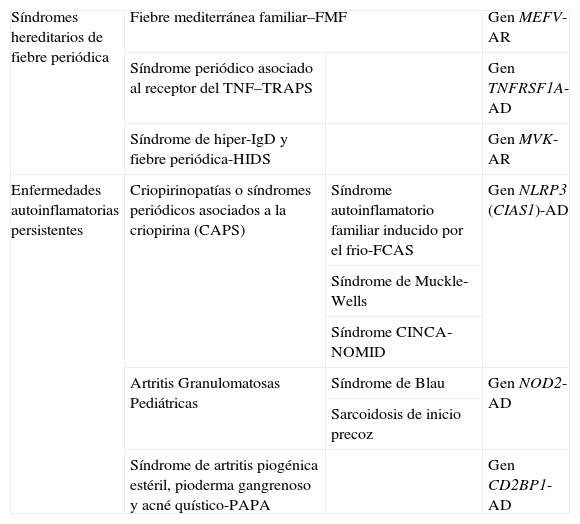
Desde su definición, el número de enfermedades autoinflamatorias hereditarias ha ido aumentando lentamente, debido a un mayor conocimiento de las mismas y a los avances de la genética3. Si bien existen diferentes clasificaciones, en la presente revisión emplearemos aquella que tiene por criterio la periodicidad o persistencia del proceso inflamatorio subyacente, diferenciando claramente 2 grandes grupos: los síndromes hereditarios de fiebre periódica y las enfermedades autoinflamatorias hereditarias persistentes (ver tabla 1).
Clasificación de las enfermedades autoinflamatorias sistémicas hereditarias, genes responsables y tipo de herencia
| Síndromes hereditarios de fiebre periódica | Fiebre mediterránea familiar–FMF | Gen MEFV-AR | |
| Síndrome periódico asociado al receptor del TNF–TRAPS | Gen TNFRSF1A-AD | ||
| Síndrome de hiper-IgD y fiebre periódica-HIDS | Gen MVK-AR | ||
| Enfermedades autoinflamatorias persistentes | Criopirinopatías o síndromes periódicos asociados a la criopirina (CAPS) | Síndrome autoinflamatorio familiar inducido por el frio-FCAS | Gen NLRP3 (CIAS1)-AD |
| Síndrome de Muckle-Wells | |||
| Síndrome CINCA-NOMID | |||
| Artritis Granulomatosas Pediátricas | Síndrome de Blau | Gen NOD2-AD | |
| Sarcoidosis de inicio precoz | |||
| Síndrome de artritis piogénica estéril, pioderma gangrenoso y acné quístico-PAPA | Gen CD2BP1-AD | ||
AD: autosómica dominante; AR: autosómica recesiva.
Antes de entrar en detalle en ellas, creemos conveniente comentar una serie de premisas válidas para todas ellas. Debido a su baja prevalencia, deben ser consideradas como enfermedades raras (menos de 5 casos/10.000 habitantes, según criterio de la UE), y consecuentemente, es posible que su difusión dentro de la comunidad médica sea limitada. Por otro lado, a pesar de tratarse de enfermedades hereditarias, la presencia de antecedentes familiares de la enfermedad suele ser baja (≈ 10% de los casos), hecho que va a dificultar su diagnóstico definitivo. Finalmente, no existen marcadores de laboratorio específicos para cada una de estas enfermedades, con la excepción de las pruebas genéticas. Por todo ello, en la practica clínica diaria, es difícil alcanzar su diagnóstico definitivo, pudiendo observarse retrasos más o menos importantes desde el debut de la enfermedad hasta su diagnóstico, la realización de múltiples pruebas complementarias y la aparición de complicaciones por el curso natural de la enfermedad.
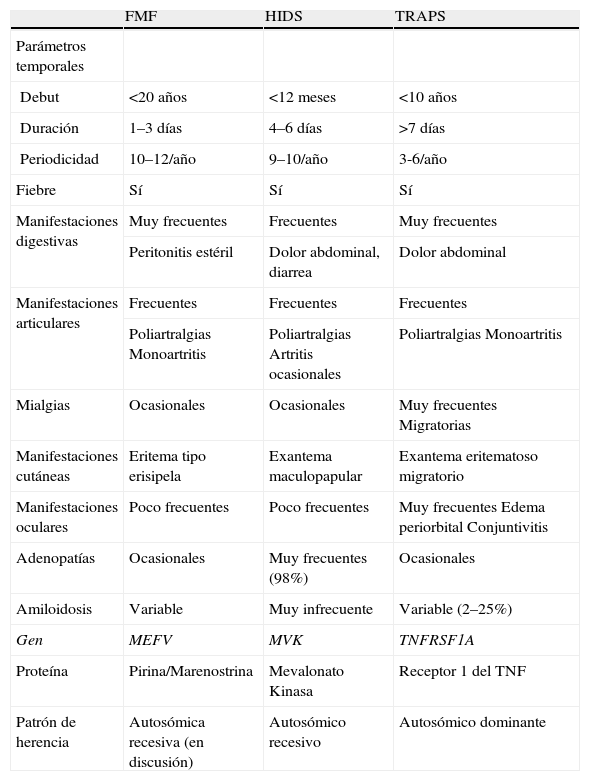
Síndromes hereditarios de fiebre periódicaEn este grupo se engloban un conjunto de enfermedades caracterizadas por la aparición de episodios inflamatorios agudos, autolimitados, de duración variable y recurrentes de forma periódica4. Se pueden identificar en ellas unos parámetros temporales (edad de inicio y duración y periodicidad de los episodios) sumamente útiles para su diagnóstico diferencial. Las principales enfermedades de este grupo son la Fiebre Mediterránea Familiar (FMF), el síndrome periódico asociado al receptor 1 del TNF (TRAPS) y el síndrome de hiper-IgD y fiebre periódica (HIDS) (tabla 2).
Principales características de los síndromes hereditarios de fiebre periódica
| FMF | HIDS | TRAPS | |
| Parámetros temporales | |||
| Debut | <20 años | <12 meses | <10 años |
| Duración | 1–3 días | 4–6 días | >7 días |
| Periodicidad | 10–12/año | 9–10/año | 3-6/año |
| Fiebre | Sí | Sí | Sí |
| Manifestaciones digestivas | Muy frecuentes | Frecuentes | Muy frecuentes |
| Peritonitis estéril | Dolor abdominal, diarrea | Dolor abdominal | |
| Manifestaciones articulares | Frecuentes | Frecuentes | Frecuentes |
| Poliartralgias Monoartritis | Poliartralgias Artritis ocasionales | Poliartralgias Monoartritis | |
| Mialgias | Ocasionales | Ocasionales | Muy frecuentes Migratorias |
| Manifestaciones cutáneas | Eritema tipo erisipela | Exantema maculopapular | Exantema eritematoso migratorio |
| Manifestaciones oculares | Poco frecuentes | Poco frecuentes | Muy frecuentes Edema periorbital Conjuntivitis |
| Adenopatías | Ocasionales | Muy frecuentes (98%) | Ocasionales |
| Amiloidosis | Variable | Muy infrecuente | Variable (2–25%) |
| Gen | MEFV | MVK | TNFRSF1A |
| Proteína | Pirina/Marenostrina | Mevalonato Kinasa | Receptor 1 del TNF |
| Patrón de herencia | Autosómica recesiva (en discusión) | Autosómico recesivo | Autosómico dominante |
La FMF es la enfermedad autoinflamatoria hereditaria más frecuente a nivel mundial. Afecta fundamentalmente a poblaciones ribereñas del Mediterráneo, con una elevada incidencia en determinadas poblaciones de su cuenca oriental (turcos, armenios, judíos y árabes)4–6. No existen datos sobre su incidencia real en nuestro país. No obstante, su mayor difusión entre la comunidad médica y la disponibilidad de análisis genéticos para su diagnóstico definitivo han permitido la identificación de múltiples casos en nuestro país durante la última década.
Las primeras descripciones de la FMF datan de inicios del siglo XX, observándose ya que es una enfermedad con episodios inflamatorios agudos, de duración breve (48–72h), que recurren periódicamente cada 3–5 semanas, si bien con cierta variabilidad de unos pacientes a otros. Las principales manifestaciones clínicas son: 1) fiebre (96%); 2) serositis inflamatoria aséptica, siendo el peritoneo y la pleura las serosas más frecuentemente afectadas (92% y 57% respectivamente); 3) manifestaciones músculo esqueléticas, como poliartralgias, polimialgias y menos frecuentemente artritis; 4) manifestaciones cutáneas, y 5) una intensa reacción de fase aguda2,4–6. Durante los intervalos intercrisis, los pacientes pueden estar totalmente libres de síntomas o presentar algunos de ellos de forma menos intensa.
Como consecuencia de estos episodios inflamatorios recurrentes, no controlados a lo largo de los años, algunos pacientes desarrollan síntomas clínicos por el depósito de la proteína amiloidea en diferentes órganos (amiloidosis secundaria), siendo el riñón el más frecuentemente afectado, presentándose habitualmente como una insuficiencia renal crónica5,7. Curiosamente, antes del advenimiento de la colchicina, el fallo renal en edades tempranas (3.a–4.a década) era una de las principales causas de mortalidad de los pacientes con FMF. En la actualidad, el tratamiento de elección es la colchicina v.o., resultando eficaz para el control total o parcial de los episodios en un 85–95% de los casos8–10.
Desde antiguo se tenía la noción que la FMF era una enfermedad transmisible a la descendencia, y clásicamente se le asignó un patrón de herencia autosómico recesivo. En 1997, 2 grupos internacionales descubrieron su base genética, al identificar mutaciones causantes de la enfermedad en un gen nuevo que denominaron MEFV (por MEditerranean FeVer), que codifica para la proteína pirina/marenostrina11,12. Se desconocen muchas de sus funciones, pero diferentes investigaciones apuntan a un posible papel como regulador negativo del inflamasoma, un complejo mutiproteico encargado de generar la forma activa de caspasa 1 y de las citocinas proinflamatorias IL-1beta, IL-18 e IL-3313.
Desde ese año 1997, el análisis mutacional del gen MEFV se ha consolidado como la prueba diagnóstica definitiva para la FMF, permitiendo diferenciarla de otras enfermedades autoinflamatorias y ofrecer un adecuado consejo genético. Sin embargo, una de las consecuencias de estos estudios es la reconsideración de su patrón de herencia, actualmente en debate, debido a que hasta un 40% de pacientes diagnosticados clínicamente de FMF en los países occidentales son portadores de un solo alelo mutado, resultado que sería compatible con un patrón de herencia dominante14.
Síndrome periódico asociado al receptor de TNF (TRAPS)La primera descripción de esta enfermedad data de 1982, cuando se da a conocer una gran familia irlandesa escocesa con múltiples miembros afectos de un síndrome hereditario de fiebre periódica. La enfermedad presentaba ciertas semejanzas con la FMF, pero existían evidentes diferencias, tales como un claro patrón hereditario dominante y episodios inflamatorios muy prolongados, de hasta varias semanas. Por todo ello, para contraponerla a la FMF, el primer nombre que recibió fue el de Fiebre Hiberniana Familiar (FHF)15. Posteriormente, se fueron describiendo nuevos casos, tanto familiares como esporádicos, que recibieron nombres diferentes tales como Fiebre Periódica Benigna, FMF dominante y Fiebre Periódica Dominante con amiloidosis16,17.
Desde un punto de vista clínico la enfermedad debuta en edad pediátrica (por debajo de los 10 años) y presenta episodios agudos prolongados (1–4 semanas) que recurren periódicamente cada 3–4 meses. Las principales manifestaciones clínicas son: 1) fiebre; 2) mialgias migratorias, debidas a una fascitis inflamatoria; 3) exantema cutáneo migratorio, centrífugo, localizado en las áreas cutáneas superficiales a los grupos musculares afectos por la fascitis; 4) serositis inflamatoria aséptica, siendo el peritoneo la serosa más afectada (92%); 5) manifestaciones oculares, tales como edema periorbital y conjuntivitis, y 6) una intensa reacción de fase aguda18. A semejanza de la FMF, la amiloidosis secundaria es su principal complicación, apareciendo con una prevalencia mayor que en la FMF (hasta en el 25% de los casos)18. Curiosamente, el principal factor de riesgo para dicha complicación parece ser el tipo de mutación responsable de la enfermedad.
Desde la primera descripción del síndrome, quedó claramente establecido un patrón de herencia autosómico dominante. En 1999 fue descubierta su base genética al identificarse mutaciones causantes de enfermedad en el gen TNFRSF1A, que codifica para el receptor 1 del TNF (también denominado p55 y CD120a). Para intentar unificar los diferentes nombres dados a la enfermedad y asociarla con su base fisiopatológica, en 1999 se propuso el acrónimo TRAPS (TNF Receptor-Associated Periodic Syndrome) para designarla1. Desde estonces han sido reportadas más de 50 mutaciones causantes de enfermedad, disponibles en la base de datos mutacional INFEVERS (dirección electrónica: http://fmf.igh.cnrs.fr/ISSAID/infevers)19.
Hasta 1999, el principal tratamiento del síndrome TRAPS eran los corticoides, generalmente a dosis elevadas y durante periodos prolongados18. Los efectos secundarios de dicho tratamiento, asociado a la edad pediátrica de muchos de los pacientes, obligaban a la búsqueda de alternativas terapéuticas. La identificación de la base molecular de la enfermedad en uno de los receptores del TNF posibilitó el empleo de agentes bloqueantes del TNF (etanercept), con resultados clínicos muy satisfactorios18,20. No obstante, con estos fármacos se ha observado una disociación entre la repuesta clínica (generalmente muy buena) y la respuesta bioquímica (muy variable, con oscilaciones importantes de los parámetros inflamatorios)21,22. Por todo ello, para intentar obtener buenas respuestas clínicas y bioquímicas, y disminuir consecuentemente el riesgo de amiloidosis, se ha empleado recientemente y con éxito el bloqueante de la IL-1 anakinra23,24.
Síndrome de Hiper-IgD y fiebre periódica (HIDS)Conocido también como Fiebre Holandesa, este síndrome fue descrito en 1984 por el Dr. van der Meer25. En la actualidad hay más de 200 casos identificados en el registro internacional de la enfermedad (dirección electrónica: http://www.hids.net). Si bien al principio la mayoría de los pacientes eran centroeuropeos, especialmente holandeses y franceses, en la actualidad han sido identificados múltiples casos en poblaciones no centroeuropeas.
Clínicamente, la enfermedad debuta a edades muy tempranas (por debajo de los 12 meses) y presenta episodios inflamatorios agudos de duración intermedia (5–6 días), que recurren periódicamente cada 5–6 semanas. Curiosamente, las inmunizaciones del calendario vacunal se identifican como factores desencadenantes de los episodios agudos. Las principales manifestaciones clínicas son: 1) fiebre; 2) linfadenopatías inflamatorias laterocervicales, bilaterales; 3) aftas orales; 4) exantema cutáneo; 5) serositis inflamatoria aséptica; 6) una reacción de fase aguda; 7) un incremento policlonal de IgD e IgA, y 8) incremento de la excreción urinaria de acido mevalónico durante los episodios agudos, no en el intervalo intercrisis26,27. A diferencia de lo comentado para la FMF y el síndrome TRAPS, la amiloidosis secundaria no es una complicación frecuente de este síndrome, habiendo sido descritos solo 3 casos hasta la actualidad28,29.
El síndrome HIDS presenta un patrón de herencia autosómico recesivo, y en 1999 fue descubierta su base genética al identificarse mutaciones causantes de la enfermedad en el gen MVK, que codifica para el enzima mevalonato kinasa30,31. Curiosamente, este mismo gen había sido identificado como responsable de la aciduria mevalónica, una grave metabolopatía32. En la actualidad se cree que ambas enfermedades son los extremos de un espectro continuo de gravedad. Así, la aciduria mevalónica representaría la forma más grave, como consecuencia de una perdida total y permanente de la actividad del enzima mevalonato kinasa, mientras que el HIDS sería la forma más leve, provocada por una pérdida parcial, pero no total, de la actividad del enzima33. Esta hipótesis es sustentada por la alta prevalencia en el HIDS de la mutación p.V377I, que se ha demostrado que genera una proteína con una actividad enzimática residual (3–5%)30,31,34.
Desde un punto de vista terapeútico, han sido múltiples los abordajes antiinflamatorios empleados en estos pacientes (AINE, colchicina, talidomida, inmunoglobulinas i.v., corticoides, estatinas, bloqueantes del TNF,…) existiendo por lo general respuestas muy dispares de unos pacientes a otros35–38. La reciente identificación del nexo fisiopatològico entre el síndrome HIDS y el inflamasoma y la IL-1 ha permitido la aplicación del bloqueante de la IL-1 anakinra en dicho síndrome, con resultados claramente esperanzadores39,40.
Enfermedades autoinflamatorias hereditarias persistentesDentro de este apartado se engloban un conjunto de enfermedades autoinflamatorias que cursan de una manera crónica, no episódica, pero que pueden presentar exacerbaciones. En este apartado se engloban los síndromes periódicos asociados a criopirina (CAPS), las artritis granulomatosas pediátricas y el síndrome de artritis piogénica estéril, pioderma gangrenoso y acné (PAPA).
Síndromes periódicos asociados a criopirina (CAPS) o criopirinopatíasTambién conocidos como síndromes urticariformes familiares, bajo este epígrafe se incluyen 3 enfermedades [síndrome autoinflamatorio familiar inducido por frío (FCAS), síndrome de Muckle-Wells y síndrome CINCA-NOMID], inicialmente descritas como entidades no relacionadas. Todas tienen un patrón de herencia autosómico dominante, comparten un mismo mecanismo molecular y representan diferentes grados de severidad a lo largo de un espectro.
Las primeras descripciones clínicas del síndrome FCAS datan de los años 40, y representa la forma más leve dentro de los CAPS41,42. Presenta un debut temprano, en muchas ocasiones en el nacimiento, y se caracteriza por la aparición de un exantema urticariforme tras la exposición generalizada al frío, que puede acompañarse de febrícula, disconfort abdominal, conjuntivitis y artromialgias. El síndrome de Muckle-Wells fue descrito en 1962 y representa un grado de severidad intermedio43. Presenta su debut durante la edad pediátrica y se caracteriza por la aparición de un exantema urticariforme, acompañado de fiebre recurrente, dolor abdominal, artromialgias, artritis. En épocas más tardías (3.a década) pueden aparecer las complicaciones que definen el síndrome: la amiloidosis secundaria (25% de los casos) y la sordera neurosensorial progresiva (35%). El síndrome CINCA-NOMID fue descrito a comienzos de los años 80 como una entidad reumatológica independiente y diferente de la AIJ de inicio sistémico44,45. Debuta en el periodo neonatal y se caracteriza por la presencia de un exantema urticariforme, una importante afectación articular (artritis recurrente o artropatías), una importante afectación neurológica (meningitis crónica aséptica, papiledema, convulsiones, sordera neurosensorial), fiebre recurrente y rasgos dismórficos.
En el año 2001 se descubrió la base molecular del síndrome FCAS y del de Muckle-Wells al identificarse mutaciones causantes de enfermedad en un gen nuevo, denominado entonces CIAS1 y conocido en la actualidad como NLRP346. En 2002, 2 grupos independientes identificaron mutaciones en el gen CIAS1 en pacientes con el síndrome CINCA-NOMID, estableciéndose en aquel momento el concepto de gradiente de severidad47,48. El gen NLRP3 codifica para la proteína criopirina o Nalp3, miembro de la familia de receptores citoplasmáticos Nod-like receptors (NLR), involucrados en la respuesta inmune innata. Dicha proteína forma parte del inflamasoma, un complejo citosólico multiproteico que, una vez ensamblado, tiene por objetivo generar la forma activa de caspasa-1 y esta a su vez generar la forma activa de las citocinas inflamatorias IL-1beta, IL-18 e IL-3313. En la actualidad se considera que las mutaciones responsables de los síndromes CAPS generan una criopirina hiperfuncionante, que se traduce en una producción excesiva y no controlada de las mencionadas citocinas inflamatorias48.
Debido a la diferente severidad de cada entidad, los tratamientos empleados en cada una de ellas han sido históricamente diferentes, oscilando desde antihistamínicos y medidas de protección frente al frío en el síndrome FCAS hasta corticoides a altas dosis en el síndrome CINCA-NOMID. Desde el descubrimiento del papel de la IL-1beta en la fisiopatología de los mismos, el tratamiento de elección de los mismos es el bloqueo de la IL-1, siendo el fármaco más empleado el anakinra, que es la forma recombinante humana del antagonista del receptor de IL-149–52. No obstante, existen otros compuestos bloqueantes de la IL-1, por mecanismos diferentes, en ensayos clínicos más o menos avanzados53,54.
Artritis granulomatosas pediátricasEste grupo engloba 2 enfermedades, la sarcoidosis de inicio precoz (EOS) y el síndrome de Blau (BS), descritas como entidades clínicas independientes en los años 70–8055–58. A pesar de las grandes semejanzas clínicas y anatomo patológicas que presentaban, durante más de 20 años existió un intenso debate médico sobre si eran una o 2 enfermedades, debido a que los pacientes afectos de EOS eran pacientes sin historia familiar de enfermedad (casos esporádicos), mientras que los afectos de BS presentaban historia familiar de la enfermedad, con un patrón de herencia autosómico dominante59. En el año 2005 quedó establecido que la base genética de ambas entidades era la misma, quedando resuelto dicho debate y proponiéndose el nombre de artritis granulomatosas pediátricas para englobar a ambas60,61.
Desde un punto de vista clínico la enfermedad presenta un debut temprano (<4 años) siendo las primeras manifestaciones: 1) exantema cutáneo eritematoso discretamente granular, y 2) poliartritis crónica simétrica, que afecta a grandes y pequeñas articulaciones y que se acompaña de una intensa tenosinovitis por infiltración granulomatosa de la sinovia. En el curso natural de la enfermedad pueden aparecer diferentes manifestaciones tales como: 1) uveítis, generalmente multifocal, agresiva y que constituye la principal causa de morbilidad en estos pacientes; 2) fiebre recurrente (50%); 3) infiltración granulomatosa en diferentes órganos (riñón, hígado, corazón), y 4) adenopatías. No existe afectación pulmonar en la forma de inicio, hecho que permite el diagnóstico diferencial con la sarcoidosis del adulto. Desde un punto de vista anatomo patológico se observan múltiples granulomas no caseificantes en diferentes órganos y tejidos60–62.
En el año 2001 se identificaron las mutaciones causantes del BS en el gen CARD15 (conocido en la actualidad como NOD2), que codifica para la proteína Nod2, que es un miembro de la familia NLR de receptores del sistema inmune innato63,64. En el años 2005 se identificaron mutaciones en el mismo gen CARD15 en pacientes afectos de EOS, siendo algunas de ellas las mismas que previamente habían sido descritas en el BS65,66. La única diferencia entre ambas enfermedades es que en el BS las mutaciones eran identificadas en todos los miembros afectos de una familia, mientras en la EOS las mutaciones identificadas eran mutaciones de novo, que aparecen por primera vez en el paciente, y que son responsables de la ausencia de historia familiar de enfermedad (casos esporádicos). Desde el año 2001 se han identificado más de 10 mutaciones causantes de enfermedad, siendo las más prevalentes las mutaciones que se localizan en el codon 334 de la proteína (p.R334Q y p.R334W)60–62.
El principal tratamiento de esta enfermedad ha sido la corticoterapia, a dosis elevadas y durante periodos prolongados, siendo por lo general ineficaces tratamientos antiinflamatorios menos potentes. Debido a sus efectos secundarios en pacientes pediátricos, recientemente se han empleado los agentes bloqueantes del TNF (infliximab), con resultados clínicos satisfactorios61.
Síndrome de artritis piógena estéril, pioderma gangrenoso y acné (PAPA)Descrito por vez primera en el año 1997, el síndrome PAPA es uno de los síndromes autoinflamatorios hereditarios más infrecuentes67. Presenta un patrón hereditario autosómico dominante, siendo sus principales manifestaciones las articulares y las cutáneas, las cuales se presentan en momentos diferentes de la vida. La enfermedad debuta en edades tempranas (<5 años), siendo las manifestaciones articulares las primeras en aparecer. La forma más habitual es una monoartritis recurrente, generalmente de grandes articulaciones, destructiva y con un líquido sinovial purulento y estéril. Conforme aumenta la edad, aparecen las manifestaciones cutáneas, siendo las más importantes: 1) pioderma gangrenoso, generalmente posterior a pequeños traumatismos o inyecciones, de difícil control terapeútico, y 2) acné quístico, que aparece generalmente a partir de la pubertad. Asimismo, han sido descritas otras manifestaciones clínicas menos frecuentes, como la hidradenitis supurativa67,68.
La base genética de dicha enfermedad fue descubierta en el año 2002, al identificarse mutaciones responsables de la enfermedad en el gen CD2BP1, que codifica para la proteína pstpip1 y que desempeña un papel en la regulación de la inflamación, al interaccionar físicamente con la proteína pirina/marenostrina y actuar sobre el inflamasoma69,70.
Los abordajes terapeúticos de esta enfermedad han sido diversos, siendo los más empleados clásicamente los corticoides a dosis elevadas. Desde la introducción de los agentes bloqueantes de citocinas (TNF e IL-1) han aparecido diferentes case reports demostrando las excelentes respuestas clínicas a los mismos, vislumbrándose como los posibles tratamientos de elección para esta enfermedad71–73.


