


El factor de crecimiento transformador-beta (TGF-β) es una citocina implicada en procesos celulares como hematopoyesis, proliferación, angiogénesis, diferenciación, migración y apoptosis celular. Aunque su papel en la artritis reumatoide no está bien definido, está considerada como una citocina inmunomoduladora según las condiciones del entorno. Numerosos trabajos han tratado de definir el papel del TGF-β en el desarrollo de la artritis murina en diferentes modelos de enfermedad, con resultados discordantes. De hecho, resultados recientemente publicados indican que TGF-β no desempeña un papel relevante en el modelo murino de artritis inducida con colágeno. Su implicación en la diferenciación y la funcionalidad de las diferentes poblaciones de células T también ha mostrado resultados dispares sobre su papel como inhibidor o promotor de la respuesta inflamatoria. En este trabajo se presenta una revisión sobre el papel de TGF-β en modelos animales de artritis.
Transforming growth factor-beta (TGF-β) is a cytokine with pleiotropic functions in hematopoiesis, angiogenesis, cell proliferation, differentiation, migration and apoptosis. Although its role in rheumatoid arthritis is not well defined, TGF-β activation leads to functional immunomodulatory effects according to environmental conditions. The function of TGF-β in the development of arthritis in murine models has been extensively studied with controversial results. Recent findings point to a non-relevant role for TGF-β in a mice model of collagen-induced arthritis. The study of TGF-β on T-cell responses has shown controversial results as an inhibitor or promoter of the inflammatory response. This paper presents a review of the role of TGF-β in animal models of arthritis.
La artritis reumatoide (AR) es una enfermedad autoinmune de etiología desconocida, que se caracteriza por la inflamación crónica que afecta a las articulaciones diartrodiales, de curso progresivo, y que provoca deformidad articular, discapacidad funcional y, en última instancia, disminución de la calidad y la expectativa de vida. Su patogenia se caracteriza por una alteración de la inmunidad celular y humoral, y por una alteración de los elementos celulares residentes en el tejido conectivo de la membrana sinovial (MS), que se comportan de forma seudotumoral, invadiendo y destruyendo los tejidos contiguos. Las primeras manifestaciones de la respuesta inflamatoria articular reumatoide parecen deberse a un cambio microvascular y a un incremento en el número de células del lining o hiperplasia sinovial. Estas alteraciones van acompañadas de una regulación alterada de citocinas, un aumento en el número de fibroblastos reumatoides con proliferación en exceso de células inflamatorias1, principalmente macrófagos y linfocitos, destrucción tisular y angiogénesis2. Los fibroblatos sinoviales reumatoides son células mesenquimales secretoras de citocinas proinflamatorias y factores de crecimiento angiogénico que favorecen el reclutamiento de células al espacio sinovial3,4. Mediante análisis por microarreglos se han identificado 2 subgrupos de fibroblastos en la MS reumatoide. En los tejidos más inflamados predomina un subtipo que expresa el factor de crecimiento transformador-beta (TGF-β) y el gen inducible por activina A, ambos característicos de los miofibroblastos. El otro subgrupo expresa genes regulados por el factor de crecimiento insulínico (IGF) y aparece en membranas sinoviales menos inflamadas5. Los macrófagos activados son los principales productores de citocinas inflamatorias destacando, entre ellas, el factor de necrosis tumoral alfa (TNF-α). Este, a su vez, induce la liberación de otras citocinas inflamatorias, como la interleucina (IL)-1, IL-6 o IL-8, implicadas en la angiogénesis y la proliferación de los fibroblastos sinoviales, así como la producción de factor de crecimiento plaquetario (PDGF), factor de crecimiento fibroblástico (FGF) y el TGF-β. La activación mantenida de estos tipos celulares provoca las alteraciones estructurales propias de la AR. Los macrófagos sinoviales, junto con los FS, desempeñan un papel importante en la destrucción tisular característica de la AR y constituyen la fuente principal de producción de metaloproteinasas (MMP). Este fenómeno está directamente regulado por citocinas proinflamatorias como IL-1, TNF-α y TGF-β, secretadas a su vez por los macrófagos residentes de la sinovial. Los neutrófilos son el tipo celular más abundante del líquido sinovial, y tienen capacidad para sintetizar TGF-β e IL-8, mediadores esenciales en su propio reclutamiento, estableciendo así un circuito autocrino. Además de los elementos celulares, factores solubles de la sinovial reumatoide como determinadas citocinas proinflamatorias (TNF-α, IL-1, IL-6, IL-17) participan en la osteoclastogénesis activando directamente los precursores de los osteoclastos6,7. Recientemente, se ha descubierto que los osteoclastos secretan IL-10, TGF-β e IL-6 y pueden actuar como células presentadoras de antígeno y activar células T CD4+ y CD8+8.
La angiogénesis es un proceso notablemente activo en la AR, sobre todo en fases iniciales, y está regulada por diversos mediadores proangiogénicos como TGF-β, angiopoyetina y los factores de crecimiento de placenta, FGF y de crecimiento vascular (VEGF). Estos factores activan a las células endoteliales e inducen la producción de enzimas proteolíticas que degradan la membrana basal y la matriz extracelular perivascular.
En la sinovial reumatoide se ha descrito la expresión de 4 factores de crecimiento, TGF-β, PDGF, FGF e IGF9,10. El TGF-β es una proteína homodimérica implicada en el control de múltiples procesos biológicos tales como angiogénesis, proliferación, diferenciación, migración y apoptosis celular11. Esta citocina tiene una estructura que consiste en 2 subunidades ligadas por puentes disulfuro, formando un homodímero de 25kDa. En mamíferos existen 3 isoformas de TGF-β (TGF-β1, TGF-β2 y TGF-β3) con funciones similares pero expresadas en diferentes tejidos. TGF-β3ha sido detectado principalmente en células de origen mesenquimal, indicando un papel diferente del de TGF-β1 y 2. Las células linfoides producen principalmente la isoforma TGF-β1, de ahí que tenga la función más relevante en el sistema inmunitario12, en particular en el control de la proliferación, activación y diferenciación de células T. Hasta el momento, se han descrito 3 receptores de TGF-β (TGF-βRI, TGF-βRII y TGF-βRIII), pero son los 2 primeros los mediadores de las respuestas biológicas de TGF-β113.
Papel de factor de crecimiento transformador-beta en la artritis reumatoideEl papel del TGF-β ha sido muy estudiado en diferentes modelos de enfermedad, como anomalías embrionarias, cáncer, enfermedades autoinmunes, aterosclerosis, hipertensión, osteoporosis y enfermedades fibrosantes e inflamatorias14. Numerosos estudios realizados en modelos de fibrosis han mostrado el efecto beneficioso de bloquear el TGF-β con oligonucleótidos antisentido15,16, anticuerpos específicos anti-TGF-β17,18 o péptidos sintéticos19-21, demostrando el papel de TGF-β como agente profibrótico. Otros estudios han mostrado que en las fases tardías de la progresión tumoral el TGF-β promueve la tumorogénesis e induce cambios en la diferenciación de las células tumorales epiteliales, un fenómeno conocido como transdiferenciación epitelio-mesenquimal22.
Los modelos animales genéticamente modificados demuestran el efecto modulador de TGF-β sobre la respuesta inmune. Ratones deficitarios en TGF-β2 y TGF-β3 presentan unos defectos que son letales en el desarrollo embrionario. Sin embargo, mutaciones del gen de TGF-β1 dan lugar a un desorden inflamatorio multiorgánico grave y a una necrosis tisular que, rápidamente, genera letalidad prenatal (entre las semanas 3-5 de edad)12. Asimismo, la ausencia funcional de TGF-β1 provoca una acumulación de células y citocinas proinflamatorias (TNF-α, IFN-γ, IL-1β) en los órganos linfoides, una producción incrementada de antígenos del complejo mayor de histocompatibilidad de clases i y ii y un déficit en la proliferación de células hematopoyéticas y endoteliales23. Histopatológicamente, estos ratones presentan infiltración masiva de linfocitos y macrófagos, principalmente en pulmones y corazón, aunque pueden observarse también en músculo, hígado, páncreas y cerebro, entre otros órganos24. Estos ratones presentan además anticuerpos anti-dsADN y anti-ssADN, y depósito de inmunocomplejos glomerulares. Otros experimentos han mostrado que ratones transgénicos para TGF-βRII (dnTβRII, que expresan un TGF-βRII con el dominio quinasa intracelular truncado bajo un promotor específico de células T y, por lo tanto, no señalizan a través del receptor), presentaron una mayor susceptibilidad a la artritis comparada con ratones de fenotipo salvaje, acompañada de un aumento en la producción de TNF-α e IFN-γ en las células T de nodos linfoides25.
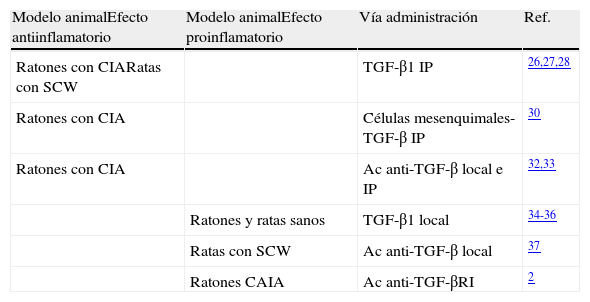
A diferencia de lo que ocurre en otras enfermedades, donde está bien definido el papel que desempeña el TGF-β, los estudios realizados en modelos de artritis han expuesto resultados muy dispares (tabla 1). Múltiples trabajos han descrito que la administración intraperitoneal de TGF-β1 reduce la incidencia y gravedad de la artritis en ratones con artritis inducida con colágeno (CIA), principalmente si la inyección se realiza en fases tardías de la enfermedad26,27. En ratas con artritis inducida por Streptococcus (SCW), la administración intraperitoneal de TGF-β en fase aguda suprime el desarrollo de artritis28. Lo mismo ocurre si la administración de TGF-β se realiza intramuscularmente29, aunque solo se ha visto este efecto si se realiza en el pico máximo de la inflamación. Un estudio muy reciente ha demostrado que células madre mesenquimales derivadas de médula ósea e inducidas con TGF-β disminuyen la incidencia y la gravedad de la artritis en ratones con CIA con la enfermedad establecida. Asimismo, incrementan la proporción Foxp3/IL-17 en el bazo y la cavidad peritoneal, y disminuyen la producción de citocinas proinflamatorias30. Otro estudio realizado en ratones con CIA mostró un aumento significativo de TGF-β1 y TGF-β2 en la articulación31, principalmente durante la fase de remisión, indicando que ambas citocinas están implicadas en la regulación de la actividad de la enfermedad. En este sentido, se ha demostrado que tanto la inyección local, como intraperitoneal, de Ac anti-TGF-β en ratones con artritis, aumenta su gravedad y los niveles de citocinas proinflamatorias32,33.
Papel del TGF-β en modelos animales de artritis
| Modelo animalEfecto antiinflamatorio | Modelo animalEfecto proinflamatorio | Vía administración | Ref. |
| Ratones con CIARatas con SCW | TGF-β1 IP | 26,27,28 | |
| Ratones con CIA | Células mesenquimales-TGF-β IP | 30 | |
| Ratones con CIA | Ac anti-TGF-β local e IP | 32,33 | |
| Ratones y ratas sanos | TGF-β1 local | 34-36 | |
| Ratas con SCW | Ac anti-TGF-β local | 37 | |
| Ratones CAIA | Ac anti-TGF-βRI | 2 |
Ac: anticuerpos; CAIA: artritis inducida con Ac anti-colágeno; CIA: artritis inducida con colágeno; TGF-β1: factor de crecimiento transformador beta 1; IP: intraperitoneal; SCW: artritis inducida por Streptococcus.
Sin embargo, también son numerosos los trabajos que exhiben un papel de TGF-β totalmente contrario. De hecho, inyecciones repetidas de TGF-β1 en la articulación de ratones sanos inducen inflamación e hiperplasia sinovial34. Igualmente, la inyección articular de TGF-β1 en ratas induce reclutamiento de neutrófilos, eritema, proliferación e infiltración sinovial, principalmente por linfocitos T35,36. Varios trabajos apoyan este concepto de TGF-β como agente proinflamatorio. El tratamiento con un Ac anti-TGF-β directamente en la articulación de ratas con artritis inducida mediante el modelo de SCW inhibe la inflamación sinovial, la resorción ósea y la producción de citocinas proinflamatorias37. Otros autores muestran que el tratamiento con un Ac-anti-TGF-βRI en ratones con artritis inducida con Ac anti-colágeno previene los signos de la artritis, el desarrollo de la hiperplasia, la inflamación y la angiogénesis sinovial2. Entre las estrategias terapéuticas descritas en la literatura para bloquear específicamente TGF-β, se encuentra p1738. Un trabajo publicado recientemente por nuestro grupo demuestra que el bloqueo selectivo de TGF-β con p17 retrasa de forma moderada los signos de artritis en el modelo murino de CIA. Sin embargo, este bloqueo no tiene efecto sobre la diferenciación y la funcionalidad de las diferentes poblaciones de células T, citocinas y grado de destrucción osteocartilaginoso, concluyendo que TGF-β no tiene un papel relevante en el modelo murino de CIA39.
El papel del TGF-β en la diferenciación de las células T murinas ha sido extensamente estudiado. En ratones, se ha demostrado que la sobreexpresión específica de TGF-β en linfocitos T conduce a la generación de células T con función reguladora y protege a ratones deficientes en IL-2 del desarrollo de inflamación sistémica grave y autoinmunidad40,41. En el modelo de CIA, la transferencia de células Treg en el momento de la inmunización reduce la gravedad de la artritis42,43. Sin embargo, el papel que desempeña el TGF-β en la diferenciación de las células Th17 no está del todo definido. Inicialmente, se describió que TGF-β e IL-6, a través de la inducción de IL-21, eran capaces de promover la diferenciación de células Tαβ CD4+ naïve a células Th17 mediante activación del factor transductor de señal STAT-3 y los factores de transcripción Rorγt y Rorα44,45. Otros estudios han mostrado que la IL-21, un miembro de la familia de citocinas IL-2, coopera con el TGF-β para inducir la diferenciación de células Th17 incluso en ausencia de IL-646,47, mientras que la IL-23, producida por células dendríticas activadas, induce la supervivencia y expansión de las células Th17 previamente diferenciadas48. Sin embargo, un estudio reciente ha demostrado que la diferenciación de las células Th17 murinas en el modelo de encefalitis alérgica experimental (EAE) puede ocurrir en ausencia de TGF-β y en presencia de IL-6, IL-23 e IL-1β49, similar a lo propuesto inicialmente en humanos. Estas células Th17 inducidas por IL-23 con fenotipo patogénico coexpresan Rorγt y T-bet y pueden dar lugar a células productoras de IL-17/IFN-γ, a diferencia de las células Th17 inducidas por TGF-β, consideradas como no patogénicas. Otros autores han propuesto que el TGF-β no actúa directamente promoviendo la diferenciación de las células Th17 murinas, sino que actúa indirectamente para regular la IL-17 por supresión de los factores que contribuyen a la diferenciación de las células Th1 y Th250. De forma paralela, muy recientemente se ha publicado que las células Th17 desarrolladas por un mecanismo dependiente de IL-23, IL-6 y TGF-β1 son patogénicas y producen elevadas cantidades de TGF-β351. Además, el TGF-β3 induce células Th17 con un papel patogénico en modelos de EAE y colitis, similar a la capacidad patogénica de las Th17 diferenciadas en presencia de IL-6 y TGF-β1, y producen cantidades más elevadas de IL-23. Por otra parte, las células TCD8+ efectoras de la sinovial reumatoide inducen la síntesis de IFN-γ e IL-17, y se ha demostrado su papel en la sinovitis crónica en el modelo de ratón K/BxN52. El TGF-β actúa en las células CD8+ induciendo la producción de Foxp3 e IL-17, dando lugar a las CD8+Treg y CD8+IL-17+, respectivamente53,54. No obstante, su papel en el desencadenamiento de CIA muestra resultados dispares55,56, indicando que estas células podrían no estar implicadas en el inicio de la enfermedad.
El papel del TGF-β en los modelos animales de AR, por tanto, no está bien definido y la discrepancia en los resultados respecto a su efecto en los diferentes tipos celulares de la sinovial reumatoide indican una alteración en los factores responsables de la vía de señal intracelular dependiente de las proteínas Smad. Estos cambios provocan respuestas anómalas del TGF-β, condicionando una alteración del comportamiento celular. Los animales Smad-3–/− presentan una respuesta inflamatoria exagerada, mientras que el déficit de Smad-2–/− es letal por defectos en el desarrollo embrionario57. Asimismo, ratones transgénicos para Smad-7 se asocian a elevada producción de citocinas por las células T helper (Th) 1 y Th2. Algunos autores han descrito que Smad-2 se ve reducida significativamente en el cartílago durante la progresión de la osteoartritis (OA) en varios modelos animales58, mientras que otros trabajos apuntan a un incremento en la expresión de TGF-βRII en fibroblastos sinoviales de pacientes con AR comparados con pacientes con OA59.
En humanos, casi todos los componentes celulares de la sinovial reumatoide pueden secretar TGF-β y, aparte de ejercer acciones sobre las células de estirpe mesenquimal, esta citocina desempeña un efecto modulador sobre las células inflamatorias. En estudios realizados in vitro con células de la sinovial reumatoide, los efectos dependientes del TGF-β son dispares, comportándose como factor inmunorregulador inductor o inhibidor de la respuesta inflamatoria según las condiciones del entorno. Entre los efectos antiinflamatorios, se encuentran la inhibición de la proliferación de linfocitos y la producción de superóxido por los macrófagos60. Sin embargo, también puede actuar como factor proinflamatorio al estimular la secreción de citocinas como TNF-α e IL-1, actuar como potente quimioatrayente de neutrófilos, activar la expresión de quimiocinas y MMP61,62, inducir la expresión de VEGF63, esencial en el desarrollo de angiogénesis en la AR, y modular la apoptosis de los FS por un mecanismo no bien definido. En este sentido, TGF-β inhibe la expresión de Fas y aumenta la de los protoncogenes Bcl-2 y Bclx, aunque otros autores apuntan a la vía de PI3K y la activación de Akt como responsables del efecto antiapoptótico de TGF-β64.
Las células mononucleares procedentes de tejido sinovial reumatoide en cultivo producen TGF-β1 y se ha demostrado su presencia en el tejido y fluido sinoviales65. Estudios de microarreglos sugieren un incremento de la vía de señalización de TGF-β en FS de pacientes con AR comparado con pacientes con OA, en concreto, TGF-β1 y su receptor (TGF-βRI), acompañado de una expresión aumentada de MMP. Este incremento de TGF-β1 se ha asociado directamente con marcadores clínicos de actividad de la enfermedad, como los niveles séricos de proteína C reactiva y los criterios de actividad ACR66, indicando una correlación directa entre el TGF-β1 y la inflamación. En este mismo sentido, se ha descrito activación de la vía de señalización de TGF-β en las células mononucleares de los agregados inflamatorios reumatoides, indicando una posible implicación patogénica de esta citocina en el desarrollo de la enfermedad39.
La MS en la AR contiene gran cantidad de células T67. Sin embargo, ha sido difícil definir el papel que estas desempeñan en el mantenimiento y la propagación de la inflamación articular. La utilización en el tratamiento de la AR de una proteína de fusión CTLA-4 humano que inhibe la coestimulación T vía CD28 ha proporcionado una prueba indirecta pero contundente de la importancia de los linfocitos T en la AR68. El papel de TGF-β en la diferenciación de las diferentes poblaciones de células T humanas es múltiple. Por un lado, se ha demostrado que la señalización a través de TGF-β1 protege a las células T reguladoras (Treg) de la apoptosis69 y es absolutamente necesaria para inducir la expresión de Foxp3, ya que se ha demostrado que células TCD4+ deficientes en TGF-β1 no pueden generar células Treg in vivo e in vitro70-73. Las células Treg pueden desarrollarse a partir de precursores tímicos de células TCD4+ en presencia de TGF-β e IL-2, dando lugar a células Treg naturales. En la periferia, las células TCD4+ naïve pueden convertirse en células Treg inducibles a través de la señalización de STAT-5 en presencia de TGF-β, aumentando la expresión del factor Foxp3 (fig. 1). La regulación del promotor de Foxp3 depende, entre otros, del factor de transcripción NF-AT y la señalización de Smad-3 inducida por TGF-β74. De hecho, el TGF-β media la expresión de Foxp3 por unión directa al promotor de Smad. Las células Treg secretan bajas cantidades de IL-2 e IFN-γ, mientras que producen elevadas cantidades de IL-10, IL-35 y TGF-β, esenciales para regular la supresión de las células T75. Recientemente, se ha descrito que, en ausencia de TGF-β1 funcional, el desarrollo de las células Treg en el cáncer podría ser producido por un mecanismo compensatorio dependiente de la expresión de TGF-β2 y TGF-β3 en el timo y la periferia76,77. Sin embargo, se desconoce si este mecanismo ocurre en la AR.

Papel de TGF-β en la diferenciación de las células T efectoras. Efecto del TFG-β y moléculas implicadas en la diferenciación y regulación de las distintas poblaciones de células T efectoras en relación con su función dentro del sistema inmunitario. IL: interleucina; TGF-β: factor de crecimiento transformador beta; Treg: T reguladora.
Clásicamente, se había asumido que la AR era una enfermedad de tipo Th1 mediada por células productoras de IFN-γ con ausencia de citocinas Th2. En la diferenciación de ambos tipos celulares, el TGF-β actúa como un regulador negativo mediante la inhibición de los factores de transcripción T-bet y GATA-3, respectivamente78,79. Sin embargo, a lo largo de los últimos 10 años, han adquirido una gran importancia las células Th17 como promotoras de la respuesta inflamatoria en la AR. En la diferenciación de las Th17 humanas, diversos mecanismos pueden estar implicados. En primer lugar, se definió que la diferenciación de las células Th17 humanas ocurría en ausencia de TGF-β80 y que la combinación de IL-1β, IL-6 e IL-23 era capaz de actuar sobre células T de memoria para producir IL-17. Sin embargo, otros trabajos exponen un papel de TGF-β, que junto con IL-21 y la expresión de Rorc2 promueven la diferenciación de TCD4+ naïve a células Th1781. En este sentido, TGF-β, a través de IL-21, suprimiría la inducción de T-bet. Los últimos trabajos apuntan a una interacción de células madre mesenquimales derivadas de médula ósea o fibroblastos sinoviales con células T como promotoras de la activación y expansión de las células Th17, las cuales podrían contribuir a la cronicidad de la AR82. Recientemente, se ha descrito un nuevo subtipo de células Th, las Th9, productoras de IL-9 e IL-10. La diferenciación de estas células a partir de células T naïve depende de TGF-β e IL-483, aunque el tratamiento de células Th2 con TGF-β también es capaz de producir células Th9. A pesar de la producción de IL-10, al no expresar Foxp3, no se consideran células reguladoras, sino células promotoras de la inflamación. De hecho, aunque su papel en la AR no ha sido investigado, en otros modelos de enfermedad inflamatoria, como la EAE, se ha señalado que pueden actuar como inductoras de la inflamación84. Asimismo, se ha demostrado que la estimulación con TGF-β e IL-1-β induce células CD4 IL-9+/IL-17+ en pacientes con diabetes autoinmune85, de ahí su posible papel en otras enfermedades autoinmunes.
ConclusiónEl TGF-β es una citocina implicada en numerosos procesos biológicos. De las 3 isoformas de TGF-β que existen en mamíferos, el TGF-β1 desempeña la función más relevante en el sistema inmunitario, en particular en el control de la proliferación, activación y diferenciación de células T. Los estudios realizados para tratar de definir el papel del TGF-β en la AR han mostrado conclusiones dispares. Las diferencias de estos hallazgos podrían deberse al modelo animal utilizado, momento de la enfermedad en el que se realiza el estudio y el protocolo empleado de inhibición de la citocina.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
FinanciaciónEste proyecto ha sido financiado por una beca de la Fundación Española de Reumatología (FER 2009) en la modalidad de ayuda a proyectos de investigación no financiados por agencias públicas. Elena Gonzalo Gil ha recibido financiación de una beca de la Fundación Española de Reumatología (FER) en la modalidad de ayuda para complementar proyectos de investigación ya financiados.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Un agradecimiento especial a los Dres. José Luis Pablos Álvarez y Gabriel Criado Carrasco, por su colaboración en el diseño de los experimentos y la revisión crítica de los resultados.


