


Vascular Ehlers-Danlos syndrome (EDS IV) is a rare genetic disorder characterised by an alteration in the COL3A1 gene which encodes type III collagen. It is the most common type of collagen in vessels of medium size and certain organs such as the intestines and the uterus. The alteration of this type of collagen produces aneurisms and ruptures of vessels and organs. A high level of clinical suspicion is required for diagnosis. It is a complex disease whose management requires a multidisciplinary team to treat the different complications that may occur.
We report the case of a 50-year-old man diagnosed with EDS IV detected incidentally after haemothorax secondary to a coughing spell.
El síndrome de Ehlers-Danlos de tipo vascular (SED IV) se trata de una rara alteración genética causada por una alteración del gen COL3A1 que codifica el colágeno tipo III. Este es el tipo de colágeno más frecuente en los vasos de mediano calibre y en algunos órganos como intestino y útero. Su alteración produce, entre otros, aneurismas y roturas de vasos y órganos. Para su diagnóstico se requiere de un alto nivel de sospecha clínica. Se trata de una enfermedad de manejo complejo que requiere de un equipo multidisciplinar para tratar las diferentes complicaciones que pueden acontecer.
Se presenta el caso de un paciente varón de 50 años diagnosticado de SED IV de forma incidental tras hemotórax secundario a acceso de tos.
Type IV or vascular (EDS IV) Ehlers-Danlos syndrome is characterised by alterations in type III collagen metabolism by mutations of the COL3A1 gene. It usually starts at a young age with spontaneous ruptures of medium-calibre arteries. Its diagnosis requires a high level of suspicion in order to correctly direct the clinical history. Its management involves a series of measures to prevent trauma or risk situations. There is no effective pharmacological treatment, only β-blockers have been tried to prevent catastrophic bleeding.
Clinical observationWe present the case of a 50-year-old male who was admitted to the pneumology department with a diagnosis of pneumonia associated with significant parapneumonic effusion; the patient reported prior symptoms of influenza, a coughing spell that triggered severe pain in the right haemothorax and presyncope symptoms.
Personal history of note included: smoker of 30 packs/year, hypertension under treatment, an episode of left carotid-cavernous fistula aged 25 treated by embolisation. At 37 years he presented with symptoms of sudden abdominal pain resulting in spontaneous haemoperitoneum secondary to rupture of the round ligament, confirmed in emergency surgery. During the surgery, splenic rupture occurred which required splenectomy.
Family history of a father who died aged 30 years due to sudden death from an unexplained case. Paternal grandmother died aged 50 from internal bleeding secondary to a ruptured axillary artery from mild trauma.
On physical examination: cutis marmorata, abundant haematomas on the upper limbs and thorax, secondary to banal trauma, slight hyperlaxity in metacarpophalangeal joints, without typical facies.
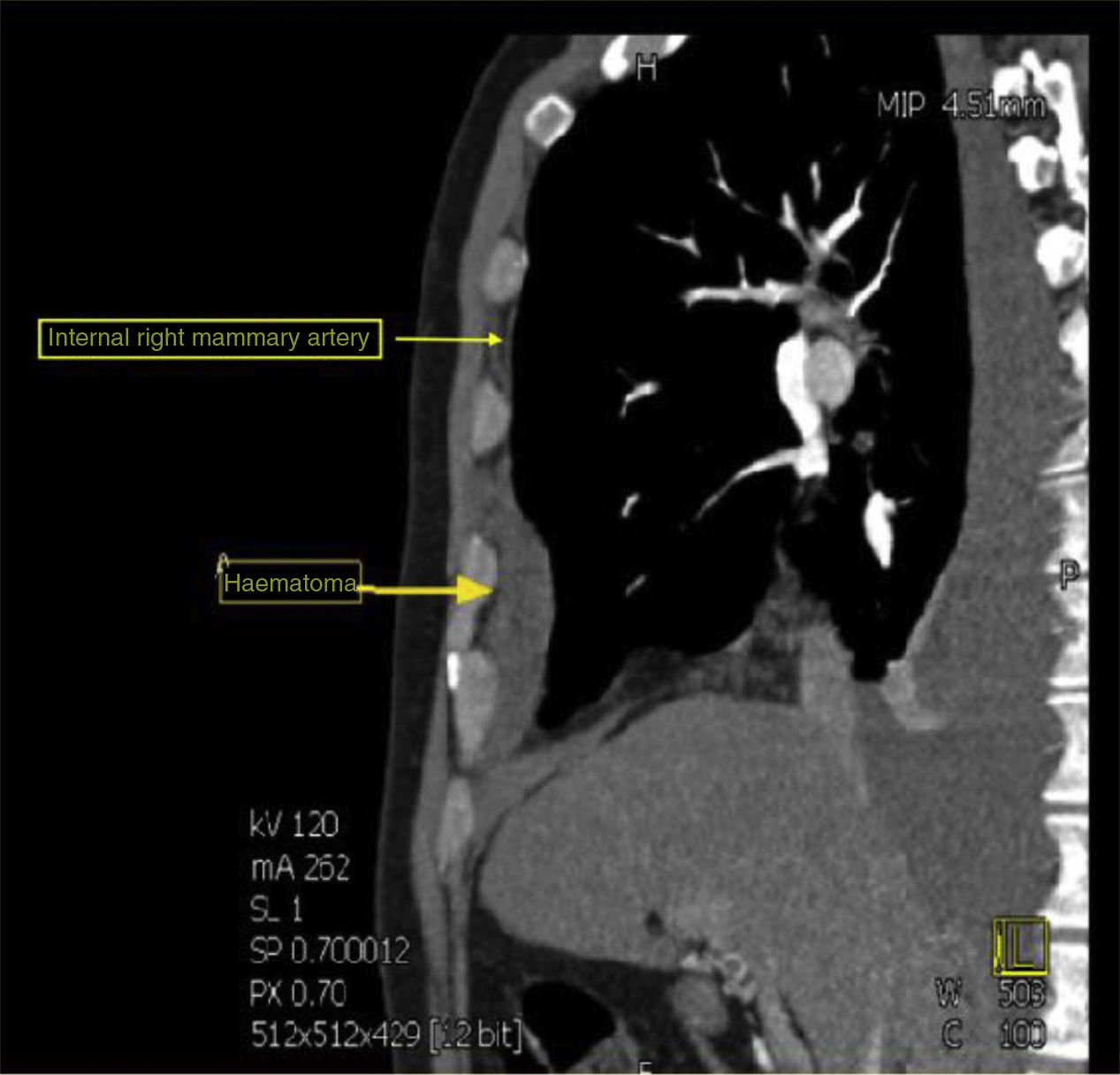
Pleural drainage of dense haematic content was performed, after which a computed tomography (CT) of the thorax was performed which revealed the presence of a ruptured aneurysm of the right internal mammary artery, causing ipsilateral haemothorax (Fig. 1). He also had an aneurysm in the common hepatic artery. Embolisation of the ruptured aneurysm was performed as treatment.

When EDS is suspected, a confirmatory genetic study is carried out for the presence of heterozygosis in the splicing variant c.636+1G>T in gene COL3A1. The variant described is not recorded on the databases consulted, but similar variants in the same point that are associated with type IV EDS are. It is transmitted with autosomal dominant inheritance. Once the suspected diagnosis has been confirmed, the study is continued and the family is given genetic counselling, and treatment is started with β-blockers and controls in outpatient clinics. After one year of follow-up, the patient remains asymptomatic with no new bleeding episodes, and under treatment with β-blockers.
DiscussionEDS is a hereditary disease caused by mutations of the collagen gene with different subtypes.1 EDS IV is the most severe form. It is inherited as an autosomal dominant trait and involves procollagen type III deficiency by mutation of the COL3A1 gene.2 It constitutes between 5% and 10% of all cases of EDS, with a prevalence of 1/50 000–1/200 000 inhabitants. It is a potentially fatal variant, increasing the risk of visceral and vascular rupture. The absence of hyperelasticity of large joints stands out, although the small distal joints of the hand may show moderate elasticity.3 Diagnosis is based on the sum of clinical criteria and is confirmed by genetic study.2 There is no effective treatment, although the administration of β-blockers has been trialled previously to reduce catastrophic bleeding.4,5
Follow-up includes minimising trauma, identifying patients and creating a care group around them, forming and facilitating individualised emergency plans, centralising treatment in referral centres, maintaining normal blood pressure and, in the event of high blood pressure, treating it strictly and monitoring the vascular tree by means of ultrasound, CT-arteriography or nuclear magnetic resonance on an annual basis.6
ConclusionsThis is a rare disease, difficult to diagnose. With regard to genetic counselling and emergency management of the patient in the emergency department, it is advisable to know the different possible manifestations to reach a quick suspicion and avoid complications. It is important to avoid trauma, and diagnostic or therapeutic vascular techniques that are not strictly necessary.
Ethical disclosuresProtection of human and animal subjectsThe authors declare that neither human nor animal testing has been carried out in this research.
Confidentiality of dataThe authors declare that they have complied with their work centre protocols for the publication of patient data.
Right to privacy and informed consentThe authors declare that no patient data appear in this article
Conflict of interestsThe authors have no conflict of interests to declare.
Please cite this article as: Álvarez K, López J, Hernández JA. Hemotórax en síndrome de Ehlers-Danlos tipo vascular. Reumatol Clin. 2019;15:e128–e129.


