



Un biosimilar (BS) es un fármaco biológico que contiene una versión de la sustancia activa de un producto biológico original ya autorizado. Los BS se comercializan al terminar el periodo de patente del fármaco original y tras demostrar que las diferencias con respecto al medicamento innovador no tienen ningún efecto relevante sobre su seguridad y su eficacia clínica. La Sociedad Española de Reumatología, en consonancia con la Agencia Europea del Medicamento, considera que por su naturaleza y complejidad de producción no se puede equiparar un BS a un genérico. La Sociedad Española de Reumatología manifiesta un compromiso inequívoco con la sostenibilidad del sistema sanitario y apoya medidas que, sin reducir la calidad asistencial, estén encaminadas a asegurar su continuidad. Por esto considera que, probablemente, la llegada de los BS mejorará el acceso de los pacientes con enfermedades reumáticas a los fármacos biológicos. Este artículo revisa la normativa de la Agencia Europea del Medicamento, el marco legal español y las controversias sobre los BS, y presenta el posicionamiento de la Sociedad Española de Reumatología sobre estos fármacos.
A biosimilar (BS) is a biological drug that contains a version of the active substance of an already authorized original biological product. The BSs are marketed after patent period of the original drug has ended and once it has been demonstrated that the differences regarding the innovative medicine have no relevant effect on its safety or clinical efficacy. The Spanish Society of Rheumatology, in line with the European Medicines Agency, considers that because of its nature and complexity of production, a BS cannot be considered to be the same as a generic drug. The Spanish Society of Rheumatology expresses an unequivocal commitment to the sustainability of the health system in our country and our steadfast alignment with all measures designed to ensure continuity, without reducing the quality of care. Therefore, we believe that the advent of BSs will likely facilitate access of patients with rheumatic diseases to the biological drugs. This article reviews the European Medicines Agency requirements for authorization, the Spanish legal framework and controversies on BS and presents the position paper of the Spanish Society of Rheumatology on these drugs.
El desarrollo, la comercialización y la disponibilidad cada vez mayor de las terapias biológicas han revolucionado el tratamiento de los pacientes con enfermedades reumáticas. Las terapias biológicas han demostrado eficacia en el control de la actividad, mejora de la función física y capacidad para modificar la evolución de estas patologías. El riesgo/beneficio y el coste/efectividad de las mismas son favorables cuando se utilizan en la población adecuada. Sin embargo, el alto precio de estos tratamientos limita su uso generalizado1. En el año 2012, dentro del top-ten de ventas de fármacos biológicos se encontraban 4 medicamentos utilizados en reumatología (adalimumab, etanercept, rituximab e infliximab)2.
En septiembre de 2013, el Comité de Medicamentos de Uso Humano (CHMP) de la Agencia Europea del Medicamento (EMA) aprobó la autorización de Remsima© e Inflectra©, fármacos biosimilares (BS) de Remicade©. Son los primeros fármacos BS de un anticuerpo monoclonal (mAb) aprobados por la EMA3,4. Asimismo, otros BS de mAb están en fases avanzadas de investigación: adalimumab y etanercept disponen de ensayos en fase 3, y otros fármacos, como rituximab y adalimumab, también ensayos clínicos en fases más tempran®as5-11.
Según la EMA12, un BS es un fármaco biológico que contiene una versión de la sustancia activa de un producto biológico original ya autorizado (fármaco de referencia). Debe demostrar similitud con el fármaco de referencia en cuanto a características de calidad, actividad biológica, seguridad y eficacia, después de un ejercicio de comparabilidad completa. Un BS no es un genérico que posee una estructura más simple y es idéntico a su fármaco de referencia.
Hasta la aprobación del BS de Remicade©, los fármacos BS aprobados por EMA eran eritropoyetinas, factores estimulantes de colonias y hormona de crecimiento, cuya estructura es muy simple si se compara con los anticuerpos monoclonales. Para su aprobación, un BS debe demostrar que la variabilidad presente y cualquier diferencia respecto al fármaco innovador no tiene efecto sobre su seguridad y eficacia. Por tanto, una vez que un BS ha sido autorizado, las agencias reguladoras garantizan que no existen diferencias significativas respecto al innovador en calidad, eficacia y seguridad.
El desarrollo por parte de la industria biotecnológica de los BS está basado en:
- a)
La expiración de las primeras patentes de terapias biológicas en el año 2001 y la introducción de la legislación europea en 2003 del concepto de BS. Estas situaciones indujeron a la industria biotecnológica a la presentación de las primeras solicitudes de BS en el año 2004, y a la aprobación en 2006 de los primeros BS13.
- b)
Factores económicos. El coste de los fármacos biológicos es elevado. En el año 2012 las ventas globales de los 10 fármacos biológicos más vendidos ascendió a más de 56.000 millones de dólares2. En este sentido, la industria biotecnológica ha encontrado en este tipo de fármacos una oportunidad de mercado.
- c)
Sostenibilidad de los sistemas sanitarios. Los gestores de los sistemas de salud ven en los BS una alternativa más económica que los fármacos de referencia y una oportunidad de ahorro de costes que contribuya a la sostenibilidad del sistema. Se estima que un BS será entre un 15-30% más barato que el fármaco de referencia.
- d)
La accesibilidad de la terapia biológica a pacientes que actualmente no pueden acceder a ella por su elevado coste.
Como algunos fármacos de referencia tienen sus patentes próximas a la expiración, distintos fabricantes están desarrollando BS, lo que podría mejorar el acceso de los pacientes a estos fármacos y disminuir costes en sistemas sanitarios como el nuestro, así como estimular una competencia que permita reducir el precio de los biológicos originales.
Normativa de la Agencia Europea del MedicamentoLos BS, según la legislación sobre medicamentos de la Unión Europea (UE), siguen un procedimiento obligado de registro centralizado que es coordinado por la EMA, y son evaluados por los mismos expertos que evalúan los productos biotecnológicos innovadores. El medicamento de referencia con el que se comparan debe estar autorizado en la UE y debe ser el mismo para todo el programa de desarrollo del BS. Un grupo de trabajo creado por el CHMP, el Biological Medicine Working Party, es el responsable de redactar las guías de medicamentos biológicos. En concreto, el CHMP se encarga de emitir una opinión científica sobre la aprobación y, por último, la Comisión Europea (CE) toma la decisión final sobre la autorización de comercialización en la UE. Este procedimiento centralizado es válido en todos los países miembros de la UE.
La EMA ha sido pionera en el desarrollo y establecimiento de un proceso regulado para la autorización de los BS. Este desarrollo se realizó por la complejidad y heterogeneidad inherente a los fármacos biológicos. El concepto de «biosimilaridad» para la fabricación y solicitud de autorización de comercialización para versiones similares de productos medicinales innovadores en la UE fue introducido en el año 2003 a través de la implementación de 2 directivas de la CE que modificaron la Directiva 2001/83 (ley europea básica que regula la autorización de medicamentos)14,15. Posteriormente, en 2005 se publicó una guía sobre los principios generales de autorización, «Guideline on similar biological medicinal products»12, que establecía los requerimientos que debía cumplir un producto para ser considerado como BS y que ha sido recientemente actualizada16. A continuación, la EMA publicó en 2006 otras 2 guías: una sobre los datos generales de producción y control, así como los requerimientos de calidad que han de cumplir los BS (Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: Quality issues)17, y otra sobre los estudios clínicos y no clínicos necesarios para demostrar comparabilidad (Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: Non-clinical & clinical issues)18. Las recomendaciones de estas guías son, en general, las mismas que se utilizan en el desarrollo preclínico de los medicamentos biológicos innovadores. Todas estas guías están siendo revisadas, sometidas a audiencia pública y actualizadas19,20. El objetivo básico del desarrollo de un BS es establecer la similitud entre el mismo y su fármaco de referencia utilizando los mejores medios disponibles para garantizar la eficacia y la seguridad demostradas previamente por su comparador.
La inmunogenicidad de los fármacos biológicos es un tema muy debatido en la comunidad científica y, con el advenimiento de los BS, el debate está en plena actualidad. Adelantándose a esta controversia, en el año 2008 la EMA aprobó una guía específica sobre inmunogenicidad, la Guideline on inmunogenicity assesment ot biotechnology-derived medical products21, que actualmente está en proceso de revisión y actualización22.
Además de las guías generales para el desarrollo de BS, la EMA ha elaborado guías específicas para el desarrollo de BS de eritropoyetinas, factores estimulantes de colonias, insulinas, hormona de crecimiento, interferón alfa y beta, folitropina y heparinas de bajo peso molecular. En diciembre de 2012 se publicó una guía específica sobre BS de mAb23 y otra guía para la valoración de la inmunogenicidad de los mAb24. El objetivo de la guía específica de BS de mAb es establecer la comparabilidad entre el BS y el producto de referencia. De este modo, se describe cómo planificar los estudios para detectar cualquier diferencia entre el BS y su producto de referencia. El proceso de comparación se realiza bajo un enfoque paso a paso y a través de un exhaustivo ejercicio de comparabilidad, en el que cualquier diferencia relevante observada debe ser justificada. Inicialmente deben realizarse estudios no clínicos cuyo primer paso son los estudios in vitro, que deben ser lo suficientemente sensibles para detectar cualquier diferencia en la actividad biológica del BS y el producto biológico de referencia. El segundo paso consiste en valorar la necesidad de llevar a cabo estudios in vivo, donde debe evaluarse si, por ejemplo, existen efectos mediados por los mAb no detectables en estudios in vitro. Sin embargo, pueden no ser necesarios si la comparabilidad de los estudios in vitro ha sido satisfactoria. El tercer paso sería la realización de estudios in vivo propiamente dichos, cuyo enfoque dependerá de la necesidad de información adicional, no siendo precisos estudios de toxicidad en humanos.
Cuando se ha completado el desarrollo no clínico se inicia el desarrollo clínico. Siempre se deben realizar estudios clínicos comparativos entre el BS y su fármaco de referencia, que, al igual que en el desarrollo no clínico, se lleva a cabo en un enfoque por pasos. En el primer paso se evalúa la farmacocinética para demostrar equivalencia farmacocinética entre ambos productos. Estos estudios deben realizarse en una población sensible y homogénea, pudiendo ser necesarias varias investigaciones en pacientes y en distintas poblaciones, sin olvidar justificar adecuadamente la elección de las mismas. Posteriormente, se realizan estudios farmacodinámicos eligiéndose marcadores farmacodinámicos para establecer comparabilidad. El segundo paso es el desarrollo clínico en el que deben realizarse: a)estudios clínicos, que deben ajustarse a las guías EMA de requerimientos clínicos de mAb y que deben demostrar comparabilidad en modelos clínicos sensibles; b)estudios de seguridad clínica, y c)plan de gestión de riesgos de farmacovigilancia (de obligado cumplimiento). Respecto a este último punto, los BS deben ser monitorizados de forma adecuada, al igual que todos los fármacos biológicos, y el solicitante de la autorización debe realizar una descripción del sistema de farmacovigilancia y elaborar un plan de gestión de riesgos, debiendo incluir los posibles riesgos debido a la inmunogenicidad de estos fármacos.
En la tabla 1 se recogen las distintas guías de la EMA sobre BS.
Principales guías EMA sobre biosimilares
| Guideline on similar biological medicinal products | CHMP/437/04 |
| Guideline on similar biological medicinal products | CHMP/437/04 Rev 1 |
| Guideline on immunogenicity assessment of biotechnology-derived medical products | EMEA/CHMP/BMWP/14327/2006 |
| Concept paper on the revision of the guideline on immunogenicity assessment of biotechnology-derived therapeutic proteins (CHMP/BMWP/42832/2005) | EMA/275542/2013 |
| Guideline on similar biological medicinal product containing biotechnology derived proteins as active substance: Non clinical and clinical issues | EMEA/CHMP/BMWP/42832/2005 |
| Guideline on similar biological medicinal product containing biotechnology derived proteins as active substance: Non clinical and clinical issues | EMEA/CHMP/BMWP/42832/2005Rev1 |
| Guideline on similar biological medicinal product containing biotechnology derived proteins as active substance: Quality issues | EMEA/CHMP/BWP/49348/2005 |
| Guideline on similar biological medicinal product containing biotechnology derived proteins as active substance: Quality issues | EMA/CHMP/BWP/247713/2012 |
| Similar biological medicinal products containing monoclonal antibodies: Non-clinical and clinical issues | EMA/CHMP/BMWP/403543/2010 |
| Immunogenicity assessment of monoclonal antibodies intended for in vivo clinical use | EMA/CHMP/BMWP/86289/2010 |
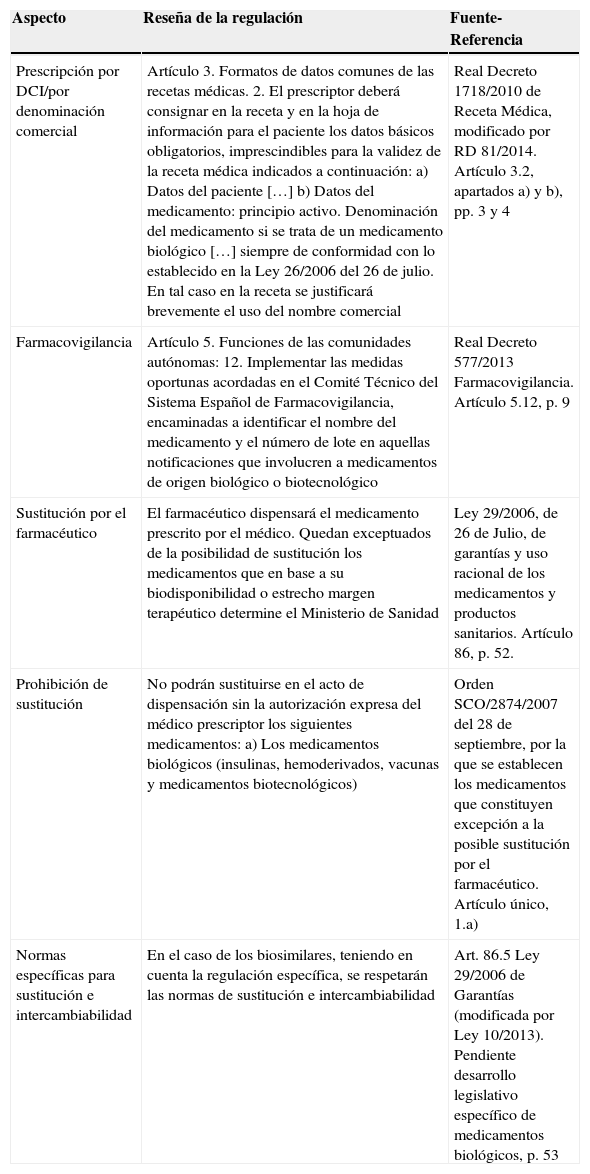
Actualmente no existe en España un marco legal específico sobre BS. Sin embargo, en la normativa vigente se encuentran distintas leyes y reales decretos en los que se abordan diversos aspectos que hacen referencia a la terapia biológica y a los BS en particular (tabla 2). A continuación se repasan estas normas.
Resumen del entorno legislativo en España sobre fármacos biológicos y biosimilares
| Aspecto | Reseña de la regulación | Fuente-Referencia |
|---|---|---|
| Prescripción por DCI/por denominación comercial | Artículo 3. Formatos de datos comunes de las recetas médicas. 2. El prescriptor deberá consignar en la receta y en la hoja de información para el paciente los datos básicos obligatorios, imprescindibles para la validez de la receta médica indicados a continuación: a) Datos del paciente […] b) Datos del medicamento: principio activo. Denominación del medicamento si se trata de un medicamento biológico […] siempre de conformidad con lo establecido en la Ley 26/2006 del 26 de julio. En tal caso en la receta se justificará brevemente el uso del nombre comercial | Real Decreto 1718/2010 de Receta Médica, modificado por RD 81/2014. Artículo 3.2, apartados a) y b), pp. 3 y 4 |
| Farmacovigilancia | Artículo 5. Funciones de las comunidades autónomas: 12. Implementar las medidas oportunas acordadas en el Comité Técnico del Sistema Español de Farmacovigilancia, encaminadas a identificar el nombre del medicamento y el número de lote en aquellas notificaciones que involucren a medicamentos de origen biológico o biotecnológico | Real Decreto 577/2013 Farmacovigilancia. Artículo 5.12, p. 9 |
| Sustitución por el farmacéutico | El farmacéutico dispensará el medicamento prescrito por el médico. Quedan exceptuados de la posibilidad de sustitución los medicamentos que en base a su biodisponibilidad o estrecho margen terapéutico determine el Ministerio de Sanidad | Ley 29/2006, de 26 de Julio, de garantías y uso racional de los medicamentos y productos sanitarios. Artículo 86, p. 52. |
| Prohibición de sustitución | No podrán sustituirse en el acto de dispensación sin la autorización expresa del médico prescriptor los siguientes medicamentos: a) Los medicamentos biológicos (insulinas, hemoderivados, vacunas y medicamentos biotecnológicos) | Orden SCO/2874/2007 del 28 de septiembre, por la que se establecen los medicamentos que constituyen excepción a la posible sustitución por el farmacéutico. Artículo único, 1.a) |
| Normas específicas para sustitución e intercambiabilidad | En el caso de los biosimilares, teniendo en cuenta la regulación específica, se respetarán las normas de sustitución e intercambiabilidad | Art. 86.5 Ley 29/2006 de Garantías (modificada por Ley 10/2013). Pendiente desarrollo legislativo específico de medicamentos biológicos, p. 53 |
En el Real Decreto 1718/2010 de Receta Médica, modificado por RD 81/2014, artículo 3.2, apartados a) y b), se indica: «Artículo 3: Formatos de datos comunes de las recetas médicas. 2. El prescriptor deberá consignar en la receta y en la hoja de información para el paciente los datos básicos obligatorios, imprescindibles para la validez de la receta médica indicados a continuación: a)Datos del paciente […] b)Datos del medicamento: Principio activo. Denominación del medicamento si se trata de un medicamento biológico […] siempre de conformidad con lo establecido en la Ley 26/2006 del 26 de julio. En tal caso en la receta se justificará brevemente el uso del nombre comercial».
Sustitución e intercambiabilidadSegún el artículo 86.4 de la Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentos y productos sanitarios, el Ministerio de Sanidad y Consumo establecerá aquellos medicamentos que, por razón de sus características de biodisponibilidad y estrecho rango terapéutico, deban constituir una excepción a los criterios generales de sustitución por el farmacéutico establecidos en el artículo 86 de dicha ley.
En la Orden SCO/2874/2007, de 28 de septiembre, se establecen los medicamentos que constituyen excepción a la posible sustitución por el farmacéutico con arreglo al artículo 86.4 de la Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentos y productos sanitarios. La relación de medicamentos afectados por esta excepción, recogida hasta ahora por la Orden de 28 de mayo de 1986, por la que se establecen los medicamentos que no pueden ser sustituidos por otros en la dispensación, requiere ser actualizada de acuerdo con los avances científicos y médicos. Con este propósito y con el objetivo primordial de asegurar la protección de la salud de los pacientes, mediante esta orden se actualiza la relación de medicamentos que, por sus características farmacológicas o terapéuticas, deben exceptuarse de las reglas generales de posible sustitución por el farmacéutico.
En su artículo único se indica que, de acuerdo con el artículo 86.4 de la Ley 29/2006 de 26 de julio, de garantías y uso racional de los medicamentos y productos sanitarios, no podrán sustituirse en el acto de dispensación sin la autorización expresa del médico prescriptor los siguientes medicamentos:
- 1.
Los medicamentos biológicos (insulinas, hemoderivados, vacunas, medicamentos biotecnológicos).
- 2.
La Agencia Española de Medicamentos y Productos Sanitarios elaborará un listado de medicamentos no sustituibles, que será de acceso público.
- 3.
Con carácter excepcional y por motivos de riesgo para la salud, el Ministerio de Sanidad y Consumo podrá establecer, mediante resolución motivada a la que se le dará la debida publicidad, que un medicamento no incluido en el apartado 1 no es sustituible en el momento de la dispensación.
Ley 10/2013, de 24 de julio, por la que se incorporan al ordenamiento jurídico español las Directivas 2010/84/UE del Parlamento Europeo y del Consejo, de 15 de diciembre de 2010, sobre farmacovigilancia, y 2011/62/UE del Parlamento Europeo y del Consejo, de 8 de junio de 2011, sobre prevención de la entrada de medicamentos falsificados en la cadena de suministro legal, y se modifica la Ley 29/2006, de 26 de julio, de garantías y uso racional de los medicamentos y productos sanitarios. Se indica que en el caso de los medicamentos BS, se respetarán las normas vigentes según regulación específica en materia de sustitución e intercambiabilidad. Actualmente está pendiente el desarrollo legislativo específico de medicamentos biológicos.
FarmacovigilanciaEn el Real Decreto 577/2013, de 27 de julio, en el artículo 2, punto 11, se indica qué medicamentos están sujetos a un seguimiento adicional. Los medicamentos incluidos en la lista que elaborará y mantendrá la Agencia Europea de Medicamentos de acuerdo con los criterios establecidos en el artículo 23 del Reglamento (CE) n.° 726/2004, del Parlamento y del Consejo, de 31 de marzo de 2004, por el que se establecen procedimientos comunitarios para la autorización y el control de los medicamentos de uso humano y veterinario y por el que se crea la Agencia Europea de Medicamentos. Dicha lista se elaborará previa consulta al Comité para la Evaluación de Riesgos en Farmacovigilancia europeo e incluirá todos los medicamentos que contengan nuevos principios activos y medicamentos biológicos, incluidos BS. La lista también podrá contener medicamentos sujetos a la obligación de realizar un estudio postautorización, o a condiciones o restricciones relativas a un uso seguro y eficaz del medicamento.
En su artículo 5.12 se indica que se deben implementar las medidas oportunas acordadas en el Comité Técnico del Sistema Español de Farmacovigilancia, encaminadas a identificar el nombre del medicamento y el número de lote en aquellas notificaciones que involucren a medicamentos de origen biológico o biotecnológico.
Recientemente se presentó una proposición no de ley en el Parlamento español (número de expediente 161/002765) sobre la regulación de la sustitución e intercambiabilidad de los medicamentos BS que fue aprobada por unanimidad con el objetivo de clarificar y actualizar su regulación. En esta proposición se recoge la necesidad de que las reglas y criterios específicos para la intercambiabilidad con otros biológicos estén recogidos en una norma de carácter general en todo el Sistema Nacional de Salud para que no se repita el debate a nivel autonómico de los «equivalentes terapéuticos», ya que en caso contrario podría menoscabarse la equidad en el acceso a los medicamentos biológicos entre distintas comunidades autónomas. En esta proposición se reclama también que, cuando se lleve a cabo la sustitución de un biológico y la incorporación de un BS en el tratamiento del paciente, se indique en su historia clínica. Asimismo, recuerda la necesidad de realizar programas de farmacovigilancia específicos para BS y recomienda que se cree un grupo de trabajo formado por representantes del Estado, las comunidades autónomas y la industria farmacéutica para abordar, precisamente, la conformación del marco legislativo citado.
Controversias sobre biosimilaresLa terapia biológica desempeña actualmente un papel muy importante en el tratamiento de las enfermedades autoinmunes y tumorales. Aunque los BS están comercializados desde el año 2006, la introducción de los mismos ha sido baja25. En este ámbito existe un amplio debate en la literatura médica sobre los riesgos y beneficios de los BS, que ha aumentado con el desarrollo de BS de mAb26-35. En particular, hay distintos aspectos que han centrado el debate, como son: la complejidad estructural de los fármacos biológicos, la seguridad, la inmunogenicidad, la extrapolación de indicaciones, la intercambiabilidad, la sustitución y prescripción por denominación común internacional (DCI o INN) o por marca comercial. En el año 2008, C. Schneider, entonces director del Biological Medicine Working Party de la EMA, ya enumeraba los pros y contras del desarrollo de los BS36. Asimismo, numerosas asociaciones científicas tanto médicas como de la industria han publicado posicionamientos sobre BS37-50.
Complejidad estructural y fabricación de fármacos biológicosHasta la reciente aprobación por la EMA del BS de Remicade©, los BS de terapias biológicas comercializados eran estructuralmente mucho más simples que los mAb. Existe una marcada diferencia entre la secuencia y la estructura de la somatotropina (191 aminoácidos y 22 daltons) con la de un anticuerpo monoclonal IgG (más de 1.000 aminoácidos y 150.000 daltons). Los mAb tienen —como todo fármaco biológico— una variabilidad inherente y una estructura compleja que no está únicamente definida por su estructura primaria (secuencia de aminoácidos), secundaria, terciaria y cuaternaria, sino también por su grado y patrón de modificaciones postraslacionales: glucosilación, metilación, fucosilación, etc. Estas modificaciones postraslacionales pueden influir en la estructura terciaria (plegamiento de unidades) y cuaternaria (configuración espacial) cambiando la integridad conformacional de los mAb y determinando diferencias en su afinidad, selectividad, actividad funcional e inmunogenicidad. La estructura general de un mAb IgG es la formada por 2 cadenas ligeras idénticas y 2 cadenas pesadas, con unas regiones constantes y otras variables. Esta estructura de los mAb puede subdividirse en región Fab (fragment antigen binding) y región Fc (fragment crystallizable). La región Fc es responsable de producir las funciones efectoras como la citotoxicidad mediada por complemento (CDC) o la citotoxicidad celular mediada por anticuerpos (ADCC); así mismo, la unión de la región Fc con células efectoras a través de receptores Fc puede producir fagocitosis o la liberación de citoquinas y compuestos citotóxicos51,52. La activación de estas funciones efectoras depende en gran parte del perfil de glucosilación del dominio Fc, por lo que la reproducción del perfil de glucosilación es un dato muy importante a tener en cuenta por los laboratorios que desarrollan BS, ya que puede tener un gran impacto en los mecanismos de acción de los mAb53.
Por otro lado, el proceso de producción de los fármacos biológicos es largo, complejo y se desarrolla en distintas etapas. El producto final depende de cada una de las etapas de dicho proceso. Por este motivo, en biotecnología se suele decir que «el proceso es el producto», y con ello se hace relación a esta complejidad en la fabricación del producto. Cualquier cambio en el proceso de producción o en las condiciones de fabricación, como el cambio en los excipientes, el uso de nuevos bancos celulares, el almacenamiento, etc., podría producir alteraciones conformacionales significativas que influyeran en la eficacia y la seguridad del producto final. Los fabricantes de BS no tienen acceso a la información del proceso de producción del fármaco de referencia, por lo que deben desarrollar su propio proceso de producción, el cual no necesariamente debe ser el mismo, y cualquier cambio debe ser evaluado de forma sistemática. Como ya se comentó en el apartado de la normativa de la EMA, el BS debe realizar un exhaustivo ejercicio de comparabilidad con su fármaco de referencia, pero a pesar de este ejercicio un BS nunca puede ser idéntico a su fármaco de referencia. Así, lo mismo se podría decir de los fármacos innovadores, ya que a lo largo de la vida de cualquier fármaco biológico el proceso de fabricación puede ser cambiado si se justifica adecuadamente54. Sin embargo, en el caso del fármaco innovador estos cambios se realizan sobre un proceso conocido y patentado, y en el caso del BS se debe desarrollar un proceso nuevo. Las variaciones a lo largo del ciclo de vida de un fármaco innovador no deben compararse con un BS producido con líneas celulares distintas y con un proceso de fabricación diferente. La prueba de que quizá no se trata de lo mismo son los requerimientos de comparabilidad que las agencias solicitan para uno u otro escenario55.
Durante el ejercicio de comparabilidad el BS debe demostrar que no existen diferencias con el fármaco innovador, siguiendo las directrices de la EMA16-18,22,23. Cualquier diferencia no justificada puede hacer que se rechace el desarrollo del mismo. Un ejemplo de esto fue la no autorización de insulinas desarrolladas como BS en las que se observaron diferencias farmacodinámicas y farmacocinéticas, y el rechazo de solicitud de Biferonex®, BS de Avonex®56, por falta de información de seguridad. Por tanto, se puede decir que un BS aprobado por la EMA ha demostrado de forma convincente que es comparable al fármaco de referencia. En este sentido, no se debería dudar de la aprobación de un BS por parte de la EMA, al igual que no se hace cuando se aprueba un nuevo fármaco biológico, lo que no quiere decir que no puedan existir cambios a lo largo de la vida de cualquier fármaco biológico, incluso su retirada por motivos de seguridad, como ya ha ocurrido en fármacos biológicos como natalizumab, mepolizumab o nimotuzumab57-59. Además, debemos establecer sistemas para garantizar su seguridad y su eficacia, al igual que se ha realizado con los fármacos innovadores.
Extrapolación de indicacionesEn las guías de EMA16-18,22,23 se recoge que si el medicamento de referencia tiene más de una indicación terapéutica, la extrapolación del perfil de eficacia y seguridad del BS en el resto de indicaciones tiene que ser justificada o, si fuese necesario, demostrada por separado para cada una de las indicaciones autorizadas. La justificación dependerá de la experiencia clínica, de los datos bibliográficos disponibles, del conocimiento del mecanismo de acción del principio activo y de los receptores involucrados en el mismo para cada indicación. Si hay evidencia de que diferentes sitios activos del medicamento de referencia o distintos receptores en las células diana están involucrados en las diferentes indicaciones terapéuticas, o de que el perfil de seguridad del producto difiere entre ellas, pueden ser necesarios datos adicionales para justificar la extrapolación de la seguridad y la eficacia de la indicación estudiada en el ensayo clínico pivotal. La decisión final estará basada en la evidencia procedente de todos los estudios de comparabilidad y en las posibles incertidumbres que aún permanezcan. Al igual que la EMA, la agencia canadiense ha desarrollado guías para regular el desarrollo de los BS60. En la guía canadiense se indica que es posible la extrapolación cuando los razonamientos aportados son suficientemente persuasivos y debe ser justificada según los mecanismos de acción, fisiopatología de la enfermedad, perfil de seguridad de las respectivas enfermedades y experiencia clínica con el fármaco de referencia. Por tanto, el laboratorio debe justificar de una manera racional su proposición de extrapolación y proporcionar los datos suficientes que la permitan.
El tema de la extrapolación es quizá uno de los asuntos que más debate y polémica ha suscitado, y son muchos los posicionamientos a favor y en contra48-50. Recientemente, la Sociedad Portuguesa de Reumatología ha publicado un documento de posicionamiento en el que se realiza una revisión exhaustiva de la evidencia donde se recogen los posicionamientos sobre extrapolación de indicaciones, así como de intercambiabilidad, sustitución e inmunogenicidad que han realizado distintas sociedades científicas, agencias de medicamentos y laboratorios36.
Hay múltiples factores que pueden influir en la extrapolación de indicaciones. En primer lugar, es conocido que los mAb pueden producir su efecto clínico a través de una gran variedad de mecanismos: bloqueo del ligando o del receptor, depleción celular (por ADCC o CDC) o por inducción de señal. La acción del mAb puede deberse a uno o varios de estos mecanismos y la participación de cada uno de estos mecanismos puede variar en cada una de las indicaciones. En concreto, infliximab y adalimumab actúan fundamentalmente a través del bloqueo del TNF-α soluble, y sin embargo en los pacientes con enfermedad inflamatoria intestinal ambos han demostrado unirse a TNF-α de membrana61. La unión al TNFα de membrana puede producir distintas acciones, como la muerte celular a través de ADCC o CDC, o apoptosis, aunque la contribución de cada uno de estos mecanismos es desconocida62,63. Debido a estos mecanismos multifactoriales de acción en pacientes con enfermedad inflamatoria intestinal, la extrapolación desde otras indicaciones en las que las funciones efectoras no parecen jugar un papel principal (por ejemplo artritis reumatoide y espondilitis anquilosante) es cuestionable. Existen además otros factores que deberían considerarse en relación con la extrapolación, como las distintas dosis y posologías en cada indicación, el uso concomitante de inmunosupresores, el uso de distintas rutas de administración para distintas indicaciones y la fisiopatología de la enfermedad.
La EMA indica que el desarrollo del BS debe realizarse en una población sensible y homogénea. Pero ¿es la artritis reumatoide el modelo más sensible? Algunos autores creen que no. Según Lee49, el modelo más sensible para detectar diferencias es la psoriasis cutánea. Según este autor, la extrapolación de indicaciones utilizando el modelo menos sensible es, por lo tanto, muy cuestionable. Asimismo indica que, incluso con el mismo mAb, los mecanismos de acción pueden ser diferentes en distintas enfermedades, y también hace referencia a los posibles problemas de seguridad e inmunogenicidad. Otros autores apuntan que a pesar de que la población elegida —artritis reumatoide— no es la más sensible, parece que la formación de anticuerpos antifármaco (ADA) es similar entre ambos fármacos y que el laboratorio ha aportado datos preliminares de un estudio observacional en 23 pacientes con enfermedad inflamatoria intestinal que sugiere una eficacia similar entre CT-P13 y Remicade©50.
La EMA, en base a los datos aportados por el laboratorio, ha aprobado el BS de infliximab (referencia EPAR) para todas las indicaciones de Remicade© (artritis reumatoide, espondilitis anquilosante, enfermedad de Crohn y colitis ulcerosa, en adultos, y en pacientes con enfermedad pediátrica, artritis psoriásica y psoriasis cutánea), teniendo en cuenta ensayos en artritis reumatoide y espondilitis anquilosante64,65. La extrapolación está basada en el amplio ejercicio de comparabilidad fisicoquímica en combinación con la demostración de equivalencia farmacocinética en enfermedades reumatológicas, obligando no obstante al laboratorio a realizar un ensayo clínico aleatorizado doble ciego entre Remsima© y Remicade© en pacientes con enfermedad de Crohn activa, así como un estudio observacional prospectivo para valorar la eficacia y la seguridad en pacientes con enfermedad de Crohn y colitis ulcerosa. En este ejercicio de comparabilidad la única diferencia objetivada entre CT-P13 y Remicade© fue un menor nivel de afucosilación, que se traduce en una menor afinidad a FcγRIIIa y, por lo tanto, una menor actividad ADCC en el modelo in vitro experimental más sensible, usando las células NK de pacientes que sufren de la enfermedad de Crohn (CD) y con genotipos de alta afinidad (VV y VF). Sin embargo, la EMA no considera estas diferencias clínicamente relevantes.
En cambio otra agencia reguladora, la Health Canada, con los mismos datos de la EMA no ha aprobado la extrapolación de indicaciones a enfermedad de Crohn, colitis ulcerosa ni a pacientes en edad pediátrica66. En el resumen de su aprobación se comenta que no se ha establecido cuál es el perfil de seguridad en pacientes con enfermedad pediátrica y que no recomiendan la extrapolación a enfermedad de Crohn y colitis ulcerosa debido a las diferencias encontradas entre el BS y su producto de referencia. No recomienda la extrapolación debido a las diferencias en los niveles de afucosilación, unión al receptor FcγRIIIa y a algunos ensayos in vitro de ADCC. En el documento se indica que el laboratorio ha proporcionado argumentos, indicando que la ADCC no es un mediador importante en la eficacia de Remicade©. Sin embargo, tras una revisión de los razonamientos aportados para la extrapolación y tras revisar la literatura, se consideró que no se puede descartar que la ADCC tenga un papel importante como mecanismo de acción en la enfermedad inflamatoria intestinal. Además, esta postura está apoyada en la observación de que certolizumab pegol, otro anti-TNF pero sin capacidad para inducir ADCC, tiene solo un beneficio marginal en la enfermedad de Crohn en comparación con otros anti-TNF. Por tanto, y ante estos datos, no se recomienda la extrapolación debido a la falta de estudios en enfermedad inflamatoria intestinal.
En resumen, en el tema de extrapolación de indicaciones hay 2 agencias reguladoras de reconocido prestigio que, con los mismos datos, han llegado a conclusiones distintas de manera razonada. ¿Cuál tiene razón? La agencia canadiense ha sido más conservadora que la EMA, pero esta última también ha recomendado la realización de ensayos clínicos y estudios observacionales en pacientes con enfermedad inflamatoria intestinal.
Sustitución e intercambiabilidadEn relación con este aspecto, la EMA afirma que las decisiones sobre intercambiabilidad y/o sustitución dependen de las autoridades competentes de cada país y están fuera del ámbito de la EMA y del CHMP. Para poder fundamentar sus decisiones, los Estados miembros disponen de acceso a la evaluación científica realizada por el CHMP y a la totalidad de la información presentada67. La agencia canadiense indica que la autorización de un BS no es una declaración de intercambiabilidad o sustitución o equivalencia terapéutica, y basa esta afirmación en posibles diferencias entre ambos fármacos, ya que no son idénticos, y en las posibles diferencias en el perfil de seguridad e inmunogenicidad de ambos medicamentos, recomendando que los clínicos participen en las decisiones de intercambio entre el fármaco de referencia y el BS68,69. Recientemente se publicó una revisión en la que se evaluaba el perfil de seguridad entre fármacos biológicos (eritropoyetinas, hormona de crecimiento y factores estimulantes de colonias). Los estudios sobre intercambio eran escasos, y más frecuentes con eritropoyetinas, sin haberse observado problemas de seguridad70. No obstante, hay que valorar estos datos con precaución, ya que la complejidad de estos fármacos no es comparable a la de los mAb.
En España, los fármacos biológicos están en la lista de medicamentos no sustituibles71. Sin embargo, en la práctica clínica habitual se cambian fármacos biológicos con frecuencia, debido a efectos adversos o ineficacia. Existen escasos datos en la literatura sobre el cambio de terapia biológica en pacientes controlados. El único estudio publicado es un ensayo clínico en pacientes con enfermedad de Crohn controlada con infliximab que son aleatorizados a seguir con infliximab o iniciar adalimumab, observándose una pérdida de eficacia y tolerancia en el grupo de adalimumab72. Existen datos preliminares presentados en el congreso ACR2013 sobre la seguridad de la sustitución entre CT-P13 y Remicade©, no habiéndose observado problemas de seguridad73,74. Asimismo, está en proyecto un estudio observacional para valorar la eficacia y la seguridad de la sustitución de infliximab por un BS en pacientes con artritis reumatoide, artritis psoriásica, espondilitis anquilosante, colitis ulcerosa, enfermedad de Crohn y psoriasis cutánea75.
La experiencia actual con los BS es relativamente limitada, por lo que hasta que mejoren las prácticas de farmacovigilancia que apoyen plenamente el intercambio de los BS parece prudente reservar al médico responsable la decisión terapéutica de sustituir un fármaco biológico por un BS en un paciente individual76.
Inmunogenicidad, seguridad y farmacovigilanciaDurante el ejercicio de comparabilidad realizado en el proceso de desarrollo del BS, la seguridad y la inmunogenicidad son aspectos clave y se tienen en cuenta a lo largo de todo el proceso de desarrollo del fármaco (ver apartado normativa EMA).
Los BS, al igual que todos los medicamentos de uso humano aprobados en la UE, están obligados a proporcionar un plan de manejo de riesgos (PMR) de acuerdo con la legislación actual de la UE sobre farmacovigilancia77. El laboratorio que solicita la aprobación del BS debe proporcionar el PMR, que debe incluir datos de seguridad para todas las indicaciones aprobadas, incluyendo seguridad a largo plazo, acciones para detectar los problemas de seguridad conocidos y desconocidos asociados al uso del fármaco, así como datos adicionales de inmunogenicidad. Los BS de Remicade© han proporcionado un RMP que fue considerado aceptable por el Pharmacovigilance Risk Assessment Committee3,4. En este PMR se detallan todas las acciones y estudios que el laboratorio fabricante realizará para un control de los potenciales problemas de seguridad. El Pharmacovigilance Risk Assessment Committee recomienda considerar la inclusión en otros registros de biológicos europeos, como el sueco y el español, aparte de los ya incluidos en el PMR aportado por el laboratorio: registro británico (BSRBR-RA) y alemán (RABBIT).
La inmunogenicidad es la capacidad para desencadenar una respuesta inmunitaria adaptativa de tipo celular y humoral. Todas las proteínas exógenas, incluyendo los mAb, tienen la capacidad potencial de desencadenar una respuesta inmune, sobre todo cuando se usan de forma repetida durante largos periodos de tiempo78,79. La incidencia de los ADA es muy variable entre los distintos fármacos y entre distintos estudios, y depende de las técnicas utilizadas para detectarlos. Las consecuencias clínicas también son variables, y ello depende de si el ADA es neutralizante o no neutralizante. Los ADA neutralizantes se unen al fármaco y evitan que desempeñe su función biológica o terapéutica disminuyendo su eficacia y asociándose a un aumento de los efectos adversos80,81. Un ejemplo clásico es la aparición de aplasia de células rojas en pacientes con insuficiencia renal tras el cambio en el proceso de fabricación de la eritropoyetina82. Existen múltiples factores que influyen en el desarrollo de inmunogenicidad. Algunos dependen del fármaco y de su proceso de fabricación: variación en la secuencia o en el plegamiento de proteínas, grado de humanización, alteración en el proceso de glucosilación, procesos de formulación, excipientes utilizados, almacenamiento, vía de administración y tiempo de tratamiento. Otros dependen del paciente: factores genéticos, edad, exposición previa a fármacos similares, enfermedad del paciente y uso de fármacos inmunosupresores asociados83. Por lo tanto, la población más sensible para detectar diferencias en la respuesta inmunogénica a mAb sería aquella en la que la inmunogenicidad no estuviese suprimida por terapias inmunosupresoras concomitantes69.
En las guías EMA sobre inmunogenicidad21,24 se discuten detalladamente los problemas de los inmunoanálisis utilizados para medir ADA, y se proporcionan recomendaciones sobre los tipos de ensayos a utilizar para valorar comparativamente el desarrollo de ADA neutralizantes y no neutralizantes. La valoración comparativa de la inmunogenicidad debería incluir no solo la incidencia de ADA, sino también los títulos de ADA y la distribución entre las poblaciones, así como su efecto en la farmacocinética, la eficacia y la seguridad69.
En el EPAR de Remsima© y de Inflectra©3,4 se afirma que el perfil de inmunogenicidad está muy bien caracterizado en los 2 ensayos clínicos llevados a cabo, y se observa que la respuesta inmune a Remicade© y su impacto sobre seguridad y eficacia son comparables al BS, por lo que estos datos pueden ser extrapolados al resto de indicaciones del producto de referencia. En el análisis de eficacia se observa que el desarrollo de ADA en ambos fármacos se asocia con un aumento en la frecuencia de reacciones de hipersensibilidad relacionadas con la infusión. En el ensayo de AR se observa que la pérdida completa de eficacia (definida como una pérdida de la respuesta ACR20) es ligeramente mayor en el BS vs. Remicade© en la semana 30 (14% vs 10%) y en la semana 54 (13% vs 11%); asimismo ocurre con la pérdida de eficacia y discontinuación de tratamiento (10% vs 65%). Sin embargo, en el estudio de EA se observa lo contrario.
Denominación común internacional o marcaLa trazabilidad de los medicamentos biológicos es un elemento de calidad en la asistencia a los pacientes, que permite asignar de forma específica a cada producto las sospechas de reacciones adversas. Una vez que un BS sale al mercado, se deben establecer sistemas adecuados que garanticen asegurar su trazabilidad. Actualmente, al BS se le asigna el mismo denominador común internacional (DCI o INN) que al innovador. Existen opiniones que indican que esta nomenclatura puede dificultar la correcta identificación del BS. Para evitar este problema, algunos expertos creen que habría que incluir en el DCI algún identificador diferencial entre BS. Otras fuentes consideran que la trazabilidad no se pierde aunque varios BS tengan el mismo DCI, ya que cada BS tiene registrado un nombre comercial diferente84.
La OMS, en su documento de «Recomendaciones para la evaluación de productos bioterapéuticos similares», indica que los BS, al igual que todos los productos bioterapéuticos, precisan un sistema adecuado que garantice la identificación específica de los mismos (es decir, su rastreabilidad). Las autoridades nacionales reguladoras deben proporcionar un marco jurídico para la supervisión adecuada de la farmacovigilancia y garantizar la capacidad de identificar cualquier producto bioterapéutico comercializado en su territorio que sea objeto de notificaciones de reacciones adversas. Todo informe de una reacción adversa de cualquier producto bioterapéutico debe incluir, además del DCI, otros indicadores importantes, tales como el nombre comercial (marca), el nombre del fabricante, el número de lote y el país de origen85.
Este es un debate abierto que debe resolverse86,87.
Posicionamiento de la Sociedad Española de Reumatología sobre biosimilaresLa Sociedad Española de Reumatología (SER) manifiesta su inequívoco compromiso con la sostenibilidad del sistema sanitario de nuestro país y se alinea con las medidas que, sin reducir la calidad asistencial, estén encaminadas a asegurar su continuidad. En esta línea, la SER considera que probablemente la llegada de los fármacos BS va a mejorar el acceso de los pacientes reumáticos a las terapias biológicas.
En este nuevo escenario de incremento de la oferta terapéutica de biológicos, la SER considera imprescindible preservar la libertad de prescripción de los médicos que realizan la indicación de fármacos según las características y circunstancias individuales de cada paciente, sin olvidar los aspectos económicos que se derivan de dicha actuación.
Los requerimientos para la comercialización de fármacos BS son muy estrictos y están muy armonizados entre las principales agencias reguladoras, lo que garantiza que la autorización de un BS se basa en la demostración de que las diferencias con respecto al medicamento innovador no tienen ningún efecto relevante sobre la seguridad y la eficacia clínica del producto.
La SER, en consonancia con la EMA, considera que no se puede equiparar un BS a un genérico. Mientras que un fármaco genérico es una copia química exacta de su fármaco modelo, el BS puede mostrar diferencias potencialmente relevantes en su estructura respecto al fármaco innovador debido a que sus procesos de producción no son idénticos.
La SER quiere poner de manifiesto sobre los fármacos BS que:
- 1.
Un fármaco BS es un fármaco biológico que es producido según las exigencias específicas de la EMA y debe demostrar similitud con su fármaco de referencia en cuanto a calidad, actividad biológica, seguridad y eficacia, en el marco de ensayos clínicos de comparación directa aleatorizados doble ciego.
- 2.
La elección de un fármaco innovador o su BS es responsabilidad exclusiva del médico prescriptor.
- 3.
Los fármacos BS no son genéricos de sus fármacos de referencia, por lo que no son sustituibles. El intercambio de un biológico por su BS es un acto médico que debe ser realizado exclusivamente por el médico prescriptor, con el consentimiento del paciente.
- 4.
La SER entiende que para una asistencia de máxima calidad las instituciones hospitalarias deben garantizar que todos los fármacos biológicos y BS que estén financiados por las autoridades sanitarias de nuestro país para el manejo de las enfermedades reumáticas deben estar disponibles en todos los hospitales del Sistema Nacional de Salud.
- 5.
Ya que los fármacos BS están sujetos a un seguimiento de seguridad igual que el de sus fármacos de referencia, es necesario crear registros de farmacovigilancia específicos. La SER tiene amplia experiencia en estos registros y se ofrece para llevar a cabo estos estudios de seguridad.
- 6.
La trazabilidad de los medicamentos biológicos es un elemento de calidad que permite asignar de forma específica a cada lote y producto las sospechas de reacciones adversas. Actualmente al BS se le asigna el mismo DCI que al innovador, por lo que la prescripción debe realizarse por marca comercial con la finalidad de conseguir una trazabilidad adecuada.
- 7.
En el caso de que el fármaco biológico de referencia tenga más de una indicación, la extrapolación de indicaciones debe justificarse según los estándares de la EMA, y en caso de ser necesario, demostrarse individualmente para cada indicación autorizada mediante ensayos clínicos de comparación directa aleatorizados doble ciego con el fármaco de referencia. La demostración de eficacia y seguridad de un BS para una indicación determinada puede no ser la misma que para una segunda indicación en la que el fármaco biológico de referencia ha demostrado eficacia y seguridad.
- 8.
El uso óptimo de los BS requiere diálogo e interacción continuo entre médicos, farmacólogos y entidades reguladoras, con la intención de preservar el derecho a la salud de los pacientes, con el objetivo de ofertarles productos de calidad eficaces y seguros.
- 9.
Este posicionamiento de la SER se actualizará periódicamente a la luz de nuevas evidencias, estimándose la próxima dentro de 2años.
Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesMiguel Ángel Abad Hernández ha recibido honorarios por conferencias de Abbvie, MSD, Pfizer y Roche, y por asesorías científicas de MSD y Abbvie.
José Luis Andreu ha recibido honorarios por conferencias de Abbvie, MSD, Pfizer, Roche y UCB, y por asesorías científicas de Abbvie, Sanofi y UCB.
Miguel A. Caracuel Ruiz no tiene conflictos de intereses.
Miguel Belmonte Serrano ha recibido honorarios por conferencias de Abbvie, MSD, Pfizer, Roche y UCB, y por asesorías científicas de MSD.
Federico Díaz-González ha recibido honorarios por ponencias (Abbvie, MSD, BMS, Celgene), por asesoría científica (Celgene, Hospira, Lilly) y por proyectos de investigación (MSD, Abbvie, Pfizer, Roche).
José Vicente Moreno Muelas ha recibido honorarios por congresos/simposios con Abbvie, Pfizer, Gebro, MSD, UCB.


