





Anti-phospatidylserine/prothrombin (aPS/PT) antibodies have been described in cutaneous Polyarteritis Nodosa (PAN) in association with specific manifestations.
ObjectivesTo determine aPS/PT antibodies in patients with PAN and its correlation with clinical manifestations.
MethodsCross-sectional comparative study including PAN patients and 20 controls (10 Microscopic Polyangiitis [MPA] and 10 Behçet’s disease [BD]). Clinical and demographic variables, treatment, serologic markers, prognosis, activity and damage indexes were evaluated. aPS/PT, anti-cardiolipin (aCL), anti-beta 2 glycoprotein 1 (anti-B2GP1) antibodies, and lupus anticoagulant (LA) were determined.
ResultsFourteen patients with PAN were included, 11 (79%) women, with disease duration of 207 months, and mostly inactive disease. Only one patient with PAN and one with BD were positive for aPS/PT IgG. LA was the most frequent antibody identified. One patient with MPA and one with BD were positive for aCL IgM; one with MPA for anti-B2GP1 IgG, and one with PAN for anti-B2GP1 IgM.
ConclusionsaPS/PT antibodies are not frequent in patients with longstanding inactive PAN.
Anticuerpos anti-fosfatidilserina/protrombina (aPS/PT) han sido descritos en Poliarteritis Nodosa (PAN) cutánea, en asociación con manifestaciones específicas.
ObjetivosDeterminar anticuerpos aPS/PT en pacientes con PAN y analizar correlación con manifestaciones clínicas.
MétodosEstudio transversal comparativo de pacientes con PAN y 20 controles (10 con Poliangeítis Microscópica [PAM] y 10 con Enfermedad de Behçet [EB]). Se evaluaron variables demográficas, clínicas, serológicas y tratamiento; índices de pronóstico, actividad y daño. Se determinaron anticuerpos aPS/PT, anti-cardiolipina (aCL), anti-beta 2 glicoproteína 1 (anti-B2GP1) y anticoagulante lúpico (AL).
ResultadosCatorce pacientes con PAN fueron incluidos, 11 (79%) mujeres, con duración de la enfermedad de 207 meses, y principalmente enfermedad inactiva. Sólo un paciente con PAN y uno con EB fueron positivos para aPS/PT IgG. El anticuerpo antifosfolípido más frecuente fue AL. Un paciente con PAM y uno con EB fueron positivos para aCL IgM; uno con PAM para anti-B2GP1 IgG, uno con PAN para anti-B2GP1 IgM.
ConclusionesLos anticuerpos aPS/PT son infrecuentes en pacientes con PAN inactiva de larga evolución.
Polyarteritis nodosa (PAN) is a necrotising vasculitis mainly affecting blood vessels of medium caliber. No specific serological tests exist for its diagnosis.1
Antiphosphatidylserine/prothrombin antibodies (aPS/PT) have been described in several different types of vasculitis. Few studies have researched the prevalence and significance of these antibodies in PAN and most have focused on the cutaneous phenotype and/or the early stages of the disease.2–4
The aim of this study was to determine aPS/PT antibodies in patients with systemic and cutaneous PAN compared with other vasculitides, determine whether there was a correlation with clinical manifestations, other antiphospholipid antibodies, activity and inflammation markers.
Material and methodsComparative cross-sectional study which included Mexicans with PAN and controls with other forms of systemic vasculitis (microscopic polyangiitis [MPA]) and Behçet’s disease [BD]). Patients were recruited from the outpatient rheumatology department from June 2018 to May 2019 in the Salvador Zubirán National Institute of medical Sciences and Nutrition. PAN diagnosis was established with the 1990 American College of Rheumatology criteria, the algorithm of the European Agency for the Evaluation of Medical Products (EMEA) and/or the Chapel Hill 20125–7 definition consensus. Patients with PAN of onset in childhood fulfilled the 2008 Ankara criteria (EULAR/PRES/PRINTO).8 MPA diagnosis was established with the EMEA algorithm and/or Chapel Hill 20126,7 consensus and BD diagnosis with criteria from the International Study Group for BD.9 Patients with hepatitis B-associated PAN were excluded, in addition to those with other autoimmune diseases, cancer, infection or treatment with anticoagulants.
Clinical and demographic variables were collected, angiography studies and diagnostic pathology. In patients with PAN diagnostic prognosis was assessed through the FFS (Five-Factor Score)10; disease activity (Birmingham Vasculitis Activity Score [BVAS 2003])11 on diagnosis and on recruitment; disease extension (Disease Extent Index [DEI])12 and damage (Vasculitis Damage Index [VDI])13 on recruitment. Serological variables were collected (erythrocyte sedimentation rate [ESR]) and reactive C protein [RCP]), and current treatment.
Anti-cardiolipin antibodies (ACA) were processed (IgG e IgM; INOVA Diagnostics; San Diego, USA) and anti-beta 2 glycoprotein 1 (anti-B2GP1) (IgG e IgM; Orgentec Diagnostika; Mainz, Germany) using automatised ELISA and considering positive values above the 99°percentile. Lupus anticoagulant (LA) was determined in plasma using the coagulometric test (LA1 screening/LA2 confirmatory; Siemens), based on the diluted Russel viper venom test. The aPS/PT antibodies were determined (IgG e IgM; INOVA Diagnostics; San Diego, USA) using the commercial ELISA kit, with reference values in keeping with the 95° percentile (IgG: ≤ 15,7 U/mL and IgM ≤ 19,5U/mL).14 The samples were preserved at -70°C and the antibodies were determined when recruitment was finalised.
The institutional ethics committee approved the protocol and participants signed an informed consent form. The procedures were performed in compliance with ethical standards and the 1964 Declaration of Helsinki.
The continuous variables were expressed as medians with an interquartile range (IQR); the categorical with absolute values and percentages. The differences between patients with PAN and control subjects were assessed using the Student’s t-test or the Mann-Whitney U test (continuous variables), and the X2 test or Fisher exact test (categorical variables). The P values <.05 were considered significant. Strata was used (Stata Corp; College Stations, Texas, USA) version 12.0 and GraphPad Prism software version 8.0.
ResultsFourteen patients with PAN were included with a median age at diagnosis of 28 years (IQR 15−42), 11 (79%) females; 10 (71%) patients had systemic PAN, four (29%) cutaneous and five (36%) of childhood onset.
On diagnosis the most frequent clinical manifestations were constitutional and musculoskeletal symptoms in 12 (86%) patients each; followed by cutaneous manifestations in 10 (71%) and peripheral neuropathy in six (43%). On diagnosis, the median of BVAS was of nine points (IQR 5–14) and the FFS of 1 (IQR 0−1). Half of the patients on whom angiography was performed presented with microaneurisms and 10/11 (91%) had a histopathological confirmation of vasculitis. No patient had a background of thrombosis. The patients with PAN presented with a median of DEI of 6 (IQR 5–8) and VDI of 1 (IQR 1–3).
Ten patients with MPA and 10 with BD were recruited as controls. Table 1 contains the demographic and medical characteristics, together with the antiphospholipid antibodies in patients and control groups. There were no differences between the groups regarding age, sex, disease duration or treatment.
Comparative analysis of patients with PAN and control group with other forms of vasculitis.
| PAN (n=14) | MPA (n=10) | Behçet (n=10) | P | |
|---|---|---|---|---|
| Male/female sex | 3/11 | 4/6 | 3/7 | .58 |
| Age at diagnosis, years | 28 (15−42) | 51 (35−61) | 36 (27−47) | .08 |
| Current age, years | 54 (25−73) | 60 (40−70) | 58 (53−63) | .62 |
| Time of evolution, months | 207 (120−362) | 118 (72−140) | 300 (228−309) | .59 |
| Current, median BVAS (min-max) | 0 (0−6) | 0 (0−18) | – | – |
| Leukocytes, cells/mm3 | 5.3 (4.2−7.3) | 5.9 (4.5−6.4) | 5.8 (4.9−7.1) | .72 |
| Haemoglobin, g/dL | 14.7 (14.1−15.5) | 14.5 (12.7−15.7) | 14.3 (14.1−15.7) | .50 |
| Reactive C protein, mg/dL | .7 (.1−.7) | .2 (0−1) | .3 (.1−.5) | .58 |
| Erythrocyte sedimentation speed, mm/h | 3 (2−9) | 6 (2−18) | 2 (2−4) | .80 |
| Creatinine, mg/dL | .6 (.6−.9) | 1.1 (.9−1.4) | .7 (.6−.8) | .06 |
| Anti-PS/PT IgG, U | 8.7 (7.2−9.8) | 8.2 (7.7−9.8) | 7.1 (6.4−7.9) | .21 |
| Positive Anti-PS/PT IgG | 1 (7) | 0 | 1 (10) | 1.00 |
| Anti-PS/PT IgM, U | 9.8 (8.4−11) | 10.7 (8.9−16.5) | 11.1 (8.3−14) | .19 |
| Positive Anti-PS/PT IgM | 0 | 2 (20) | 1 (10) | .25 |
| ACL IgG, UGPL/mL | 5.2 (4.9−5.6) | 5.3 (4.9−6.1) | 4.9 (4.8−5.6) | .91 |
| Positive ACL IgG | 0 | 0 | 0 | – |
| ACL IgM, UGPL/mL | 7.5 (6.9−9.5) | 9.3 (7−10.2) | 9 (6.8−10) | .36 |
| Positive ACL IgM | 0 | 1 (10) | 1 (10) | .50 |
| Anti-B2GP1 IgG, U/mL | 3.3 (3.2−3.5) | 3.2 (3.2−3.3) | 3.3 (3.2−3.4) | .32 |
| Positive Anti-B2GP1 IgG | 0 | 1 (10) | 0 | – |
| Anti-B2GP1 IgM U/mL | 3.5 (3.5−3.7) | 3.6 (3.4−3.8) | 4.1 (3.6−4.7) | .51 |
| Positive Anti-B2GP1 IgM | 1 (7) | 0 | 0 | – |
| Positive LA | 5 (36) | 6 (60) | 3 (30) | .45 |
| Current treatment | ||||
| PDN | 5 (36) | 5 (50) | 3 (30) | .74 |
| AZA | 3 (21) | 4 (40) | 4 (40) | .47 |
| MTX | 1 (7) | 0 | 1 (10) | 1.00 |
| CPM | 1 (7) | 0 | 1 (10) | 1.00 |
| MPM | 1 (7) | 0 | 1 (10) | 1.00 |
| Current does of PDN, median mg (min-max) | 5 (5−40) | 10 (5−25) | 5 (2.5−60) | .63 |
The scores are presented as n (%) or median (p25–p75) if not specified to the contrary.
ACA: anticardiolipin antibodies; Anti-B2GP1: anti-beta 2 glycoprotein 1 antibodies; aPS/PT: antiphosphatidylserine/prothrombin antibodies; AZA: azathioprine; BVAS: Birmingham Vasculitis Activity Score; CPM: cyclophosphamide; LA: lupus anticoagulant; MPA: microscopic polyangeiitis; PAN: poliarteritis nodosa; PDN: prednisone; MTX: methotrexate; MPM: mycophenalate mofetil.
On recruitment, patients with PAN had had a disease duration of 207 months (IQR 120−362) and the majority presented with an inactive disease (median of BVAS 0 points [IQR 0−6]), low levels of acute phase reactants (PCR .7mg/dL [IQR .1-.7] and ESR 3mm/hr [IQR 2−9]), and a median dose of prednisone of 5mg/day.
Some patients with PAN presented with persistent symptoms: constitutional in two (14%), musculoskeletel in four (29%) and peripheral neuropathy in four (29%).
One patient with PAN and one with BD tested positive for aPS/PT IgG, and none of the patients with PAN tested positive for aPS/PT IgM; however, this antibody was present in two patients with MPA and one with BD. The LA was the most common antibody, in five (36%) patients con PAN, six (60%) with MPA and three (30%) with BD (P= .45). One patient with MPA and one with BD had positive aCL IgM; one with positive MPA anti-B2GP1 IgG and one with positive PAN anti-B2GP1 IgM. Fig. 1 shows the rates of aPS/PT IgG and IgM antibodies in patients with PAN and in controls.
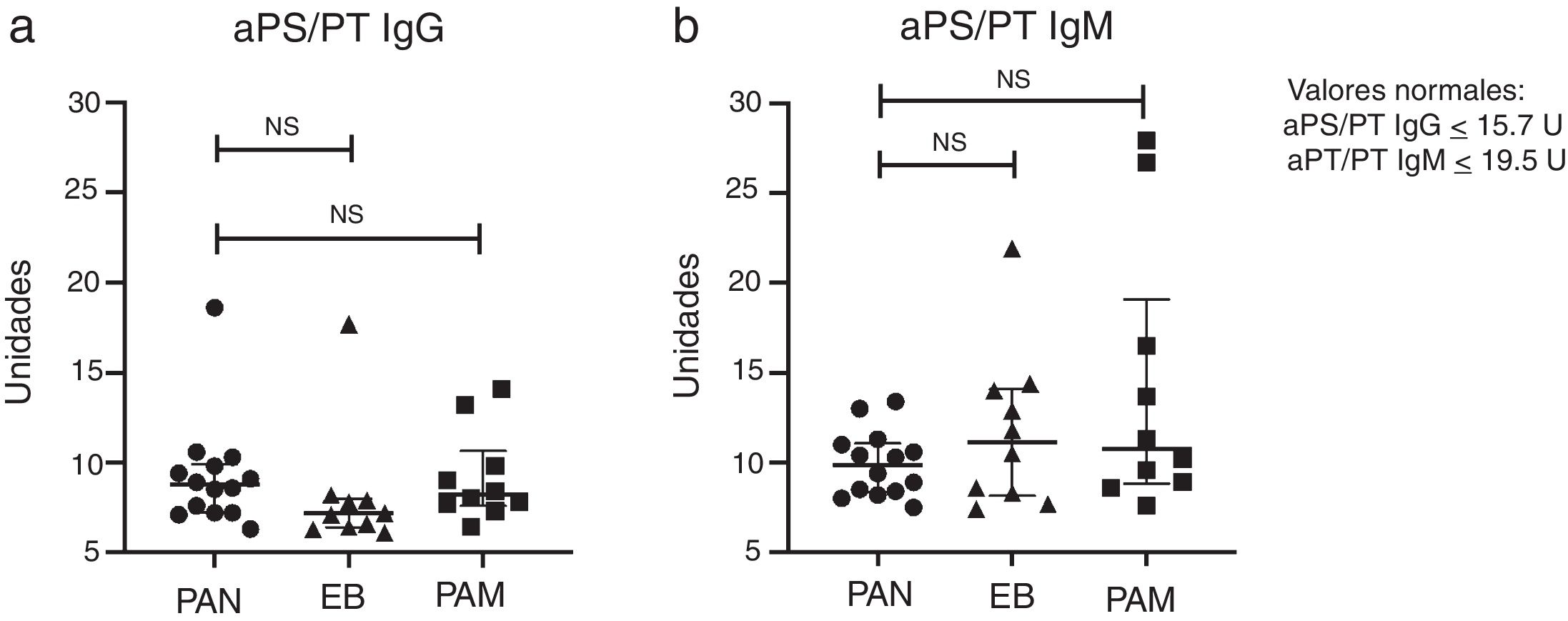
The only patient with PAN and positive aPS/PT IgG also tested positive for LA. LA diagnosis of PAN presented with multiple mononeuropathy, fever, musculoskeletal symptoms, subcutaneous nodules and livedo reticularis. At the time of recruitment, 11 months after diagnosis, they presented with constitutional and musculoskeletal symptoms in addition to neuropathy (BVAS 6 points).
DiscussionThis study analysed the presence of aPS/PT antibodies in the patient with PAN which was predominantly of a systemic phenotype and its association with onset manifestations. No patient with PAN tested positive for aPS/PT IgM and only one presented with positive aPS/PT IgG. These results contrast with previous studies where the presence of aPS/PT antibodies and other antiphospholipid antibodies were described in a patient with PAN.2–4
Kawakami et al. found that 13 out of 16 (81.3%) patients with cutaneous PAN had positive aPS/PT IgM, with higher rates than controls with generalized lupus erythematous. In this study, patients with MPA and healthy controls did not present with positivity for these antibodies; furthermore, they reported a positive correlation between aPS/PT IgM antibodies and PCR levels in patients who also tested positive for LA.2
The same group also described the correlation between cutaneous manifestations (livedo racemosa and inflammatory plaques) and the presence of LA and aCL and aPS/PT antibodies in patients with cutaneous PAN. They found higher levels of aPS/PT IgM antibodies in patients with livedo racemosa and of aPS/PT IgG and aCL IgG in those with inflammatory plaques.3
Furthermore, Okano et al., studied patients with PAN with active cutaneous manifestations and determined aPS/PT antibodies and cytokines before and after treatment with cyclophosphamide and glucocorticoids. In this study it was found that the aPS/PT antibodies and levels of IL-2 after treatment were lower compared with the levels prior to treatment.4
The above indicates that the presence of aPS/PT antibodies in patients with PAN could be conditioned by the duration of the disease, clinical phenotype, and presence of activity, since the majority of patients with reactivity for these auto antibodies presented with cutaneous, active PAN of recent onset, and were new to treatment.2,3
The patients included in this study had a prolonged disease evolution, they were predominantly of the systemic phenotype, and showed an inactive disease at the time of recruitment; also, cutaneous manifestations such as livedo reticularis or racemosa were not present, which could have explained the absence of aPS/PT in our cohort.
This study has its limitations. The sample size was small and there were no healthy controls with which to compare antibody rates. The patients had a prolonged disease duration, added to which most were in remission and had received treatment previously. Finally, the antibodies were determined on a single occasion. However, we believe that this study reflects a panorama of patients with outpatient PAN, includes patients with a predominantly systemic phenotype and as such provides additional information to previous reports on aPS/PT antibodies in cutaneous PAN. Also, it included patients with other forms of vasculitis as controls and a complete profile of antiphospholipid antibodies was determined.
To conclude, the results from this study suggest that aPS/PT are infrequent in patients with PAN of long evolution in remission. To confirm these results, longitudinal studies are required, with a larger number of patients with cutaneous and systemic phenotypes, an active disease and in remission, prior to and posterior to treatment.
Conflict of interestsThe authors have no conflict of interests to declare.
FinancingThis study did not receive any type of financing.
The authors would like to thank Isela Chan Campos, Darinel Hernández Hernández and Andrés Valencia Martínez for their valuable support in taking samples and in the determination of the lupus anticoagulant.
Please cite this article as: Sánchez-Cubías SM, Martín-Nares E, Hernández-Molina G, Nuñez-Alvarez CA, Sedano-Montoya MA, Vargas-Ruiz AG, et al. Anticuerpos antifosfatidilserina/protrombina en pacientes con poliarteritis nodosa. Reumatol Clin. 2021;17:521–524.


