



The present document is a position statement of the Mexican College of Rheumatology on the use of biosimilars in rheumatic diseases. This position considers that biosimilars should be considered as interchangeable, that automatic substitution without previous notice in stable patients during follow-up is not ethical, that the approval of a biosimilar should only be given after exhaustive review of preclinical and clinical data marked by Mexican regulations, that it should be clearly stated in the nomenclature of biologic drugs which is the innovator and which is the biosimilar, that it is not correct to choose a biosimilar as treatment based only on economic reasons or extrapolate indications based only on the approval of the innovator and in the absence of safety and efficacy data for the biosimilar.
El presente documento refleja el posicionamiento del Colegio Mexicano de Reumatología y de expertos sobre el uso de medicamentos biocomparables (conocidos como biosimilares en otros países) en enfermedades reumáticas. En resumen, este posicionamiento considera que si bien los biocomparables deben considerarse como intercambiables, no es ética la sustitución automática de medicamentos sin previo aviso en pacientes estables durante el seguimiento; que la aprobación de un biocomparable debe llevarse a cabo solo después de revisar exhaustivamente las pruebas preclínicas y clínicas señaladas por la ley mexicana; que debe modificarse la forma de enfatizar en su nomenclatura que se trata de un medicamento biotecnológico innovador o biocomparable de manera clara; que no es adecuado elegir como tratamiento un biocomparable basándose únicamente en aspectos económicos ni realizarse la extrapolación de indicaciones basándose únicamente en la aprobación obtenida por el innovador y en ausencia de datos de seguridad y eficacia para el biocomparable.
The introduction of biological drugs has produced a watershed in the history of the treatment of chronic disabling, and even potentially fatal, diseases, as occurs in oncology, where biological agents, in particular cases, are less toxic and lead to longer survival than conventional treatments; with respect to rheumatology, their success has been much more evident in diseases like rheumatoid arthritis (RA) or spondyloarthritis, in which a considerable percentage of patients achieve clinical remission, a condition that, in the past century, was attained in less than 20% of the patients.1 In addition, it is evident that patients return to work, which represents less expense for health institutions. Currently, we are faced by the expiration of the patents of certain innovative drugs and, with this, the arrival of the so-called biosimilar drugs, which in Mexico are referred to as “biocomparables”. In accordance with the established definition, a biosimilar drug is a noninnovator biological product that has been shown, by means of tests established in the Mexican regulatory framework, to be comparable to the reference biological drug in terms of safety, quality and efficacy. On the international level, these drugs are known as biosimilar medical products.2 It is worth mentioning that biosimilar medicines are in themselves biological agents and the major difference lies in the fact that the innovators launched the product and the formulation was replicated when the patent expired.
The introduction of biosimilar drugs is accompanied by the possibility of reducing the cost of antirheumatic treatments and increasing their availability when the patents of the reference products expires, based on the supposition of their lower cost.3 One of the current advantages of these products is that, once the comparability of a drug has been demonstrated, they may obtain approval from health agencies for the same therapeutic indications authorized for the innovator drug,4 provided they can make available the scientific evidence that enables the extrapolation of these indications. Biosimilars can be acquired in the European Union since 20064 (with the approval of Omnitrope®, somatropin), in the United States since 2015 (with the approval of Zarzio®, filgrastim) and in Mexico since 2014 (with the approval of Zarzio®, filgrastim).
Currently, among the therapeutic options in rheumatic diseases, there is only one biosimilar approved for marketing in Mexico; this product, referred to as CT-P13, is an infliximab biosimilar. This was accomplished by applying the existing legislation on biosimilars. The biosimilar drug is known as Remsima®; nevertheless, a number of biosimilar drugs are now under development and soon there will be more on the market. The regulatory framework to which biosimilars are subjected has undergone changes. For example, the introduction of NOM-257-SSA1-2014 concerning questions related to biotechnological medicines. According to the most recent version, approval requires the presentation of characterization data and clinical trials involving patients, which are also included in other updated regulations and reference guidelines.5 In rheumatology, certain biological drugs have encountered access barriers to their approval and prescription on the part of physicians, favored in part by the introduction of biological products authorized prior to the current Mexican regulatory framework. In light of new scientific evidence, the Mexican College of Rheumatology (CMR) presents an update of its position with respect to biosimilars, which was published for the first time in 2012.6
DefinitionsIn Mexico, as we mentioned above, biosimilars are defined as noninnovator products that have the characteristics of quality, efficacy and safety comparable to those of the reference drugs.6,7 There are other so-called biotechnological medicines,8,9 still yet to be classified, which were registered prior to the current regulatory framework for biosimilar drugs; for these cases, Norma Oficial Mexicana (Official Mexican Standards) NOM-257-SSA1-2014,5 establishes those drugs that must undergo a review of the health registration process in accordance with article 157 of the Regulation on Health Supplies, for the purpose of meeting the requirements established by the General Health Law.10
The term “comparability” refers to the technical evaluation of the quality attributes and the impact of changes in the manufacturing process in the different stages of the biotechnological development or after approval of the product. This can involve analytical and clinical testing that demonstrate that the quality, efficacy and safety are equivalent to those of the reference product.11–14 “Equivalence” refers to the fact that the biosimilar has a qualitative and quantitative composition comparable to that of the reference drug, provides the same therapeutic effect15,16 and complies with a high level of comparability with the reference product approved by the authorities.17
Clinical safety of biosimilars is not assessed only in terms of adverse events; it also involves the evaluation of the immunogenic potential, defined as the ability of a molecule to provoke a long-term adaptive cellular or humoral immune response and generate immunological memory.18,19 Aside from pharmacokinetics, pharmacodynamics, safety and efficacy, which are evaluated in the majority of clinical trials with biosimilars to be employed in the field of rheumatology, there are other characteristics of biosimilar drugs that should be taken into account to establish a stance on their use. Here we provide their definition.
Traceability is understood to be the ability to reconstruct the history, route or application of a drug product, thus identifying the origin of its components, the processes it undergoes and its distribution and storage from its production and throughout its entire “life cycle”. It consists in being capable of identifying the stages that a product has gone through during the process of being manufactured (manipulations, composition, machinery employed, temperature to which it was subjected, batch number, etc.), which are considered important and can produce variations in the final product that reaches the consumer.20,21
NomenclatureAt the present time, in Mexico and other countries, innovator products and biosimilars are marketed and distributed under the same international nonproprietary name as the active ingredient. Therefore, both products share the same code assigned by the health sector. If the products are on the list of essential medicines, this administrative process, regulated by the Secretariat of Health under the General Health Council, favors that said products can be interchanged indiscriminately in order to facilitate competitive supply and demand, or availability of the drug. However, the latter is problematic in clinical terms, especially with regard to traceability, which is an indispensable aspect in the field of pharmacovigilance and should be implemented by all of the individuals or legal entities that intervene in the chain of production, marketing, distribution and dispensing of the product in question (drug companies, suppliers, logistic operators, pharmacists, physicians and nurses).20,21 Thus the United States Food and Drug Administration issued a proposal as a guidance for the nomenclature of biosimilar products that are not proprietors of the original name, and that, at the time of writing of this review, was in a stage prior to its official publication. This proposal establishes that, currently, is not appropriate for biosimilars to share the generic name or international nonproprietary name as there is a need to clearly identify biological products to make pharmacovigilance more efficient and plainly differentiate the products that have not been determined to be interchangeable.22 In Mexico, we still do not have official proposals to solve this current problem in benefit of the patient. The World Health Organization has proposed to add a series of numbers and letters to the international nonproprietary name using prefixes and suffixes to classify the molecules more accurately, but this has not been adopted universally. The European Medicines Agency (EMA) has not made it clear what information should be included in the technical specifications, although the European position is to align biosimilars and innovators in this regard and identify them by a single international nonproprietary name. Thus, the addition of a code or qualifier would be contrary to this idea. However, it is true that in other parts of the world, with markets that are less regulated, the notion has had greater approval. This idea has been promoted by the innovator industry to hinder the introduction of biosimilars in the market.
Safety and ImmunogenicityThe clinical data on safety, including evaluations of immunogenicity, are essential for assessing biosimilarity23 because of the probability of small differences between proteins and the concern that they could lead to an increase in immunogenicity and hypersensitivity. A biosimilar product can differ from the reference product in the formulation, impurities, excipients and clinically inactive components. This can result in clinically relevant immunogenic differences, important enough to mean the refusal of the license of a product that seeks approval as a biosimilar.23 Both the Food and Drug Administration and the World Health Organization require at least one immunogenicity study to compare the immune response of the biosimilar and the reference product.24–26 In addition, the EMA proposes that follow-up studies of immunogenicity and safety should be conducted after the authorization.27
Interchangeability and Automatic SubstitutionIn Mexico, the regulations do not establish that a biosimilar can be considered to be interchangeable (Regulation on Health Supplies). There is no position on the part of the Mexican regulatory agency (Federal Commission for Protection Against Health Risks [COFEPRIS]) on the interchangeability or substitution of biotechnological drugs by their corresponding biosimilars; however, the practice of automatic substitution at the institutional level is indiscriminate, partly due to a clear legal vacuum and the lack of endorsement and agreement among the different key authorities and actors in the pharmaceutical market. The expression comparability does not necessarily signify interchangeability. The term interchangeability refers to the designation that means that a biosimilar can be prescribed rather than the reference product.28 Substitution without the intervention of the health care provider who prescribed the reference product is automatic substitution.2 A biosimilar drug can be considered to be interchangeable with the reference product only when the product fulfills the definition of biosimilar and it has been shown that it has the same clinical result in the patient after changing or alternating the formulation, a fact that is assessed over a reasonable period of time.29 Interchanging the formulation (or “switching”) refers to beginning treatment with the reference product and, subsequently, continuing with the biosimilar or vice versa. Substitution refers to initiating treatment with the reference product and changing to the biosimilar, and subsequently returning to the reference product, or beginning with the biosimilar and continuing with the reference drug and finishing with the biosimilar.29
In addition, to assume interchangeability, the risk in terms of safety or a decrease in the efficacy upon switching or alternating between the reference product and the biosimilar is no greater than the risk for the patient if he or she only takes the reference drug, without changing the product or alternating between the reference product and the biosimilar.30
However, automatic substitution entails the risk of failure in terms of pharmacovigilance, since there is no medical indication involved, this being a practice carried out by the pharmacist, without adequate traceability.2
Indication ExtrapolationExtrapolation means that once the comparability has been established in one or more indications, a biosimilar can be approved for some or all of the additional indications for which the reference or innovator product is authorized, with no need for comparative clinical trials involving the remaining indications.31,32 In the United States, it is possible to authorize a biosimilar for multiple conditions for which the reference drug has been authorized based on evidence for its use for one indication, provided it is supported by a scientific justification.23 In Mexico, we do not have an official document that details the characteristics for indication extrapolation in biosimilars and this is left to the discretion of the authority, partially backed by the opinion of expert committees. On the other hand, the possibility of extrapolation of indications is one of the most attractive aspects of the development of biosimilars33; nevertheless, this is controversial since the pathophysiology of the diseases and the mechanisms of action of the biosimilars are not necessarily the same in all conditions nor with all the types of biosimilar drugs. It is worth mentioning that not every country allows indication extrapolation; one example is the EMA,27 which permits that the results of clinical trials involving patients with rheumatic diseases be extrapolated to inflammatory bowel diseases, whereas the Public Health Agency of Canada34 does not.33 The regulatory considerations for indication extrapolation are shown in Table 1.
Regulatory Considerations for Indication Extrapolation in Biosimilars.
| Health Canada34 | Authorization for a biosimilar can include all the indications approved for the biological product authorized in Canada to which a reference is made |
| The authorization of the requested indications depends on the demonstration of similarity between the biosimilar and the reference biological drug on the basis of data derived from comparative structural, functional, nonclinical and clinical studies. Where similarity has been established, indications may be granted even if clinical studies are not conducted in each indication. This requires a detailed rationale that scientifically justifies the authorization of the biosimilar in each indication, taking into account the mechanism or mechanisms of action, the pathophysiological mechanism of the disease or conditions involved, safety profile, dosing regimen, clinical experience with the reference biological drug and any case-by-case considerations. Certain conditions may warrant additional clinical data to support a particular indication. | |
| EMA27 | If biosimilarity has been demonstrated in a therapeutic indication, the extrapolation of clinical data to other indications of the reference product could be acceptable, but must be scientifically justified. In cases in which it is not clear whether the safety and efficacy confirmed in one indication would be relevant in another indication, additional data will be required. Extrapolation should be considered taking into account all of the data, that is, quality, nonclinical and clinical data. It is expected that safety and efficacy could be extrapolated if the biosimilar comparability has been demonstrated by means of exhaustive physicochemical and structural studies, as well as in vitro functional tests complemented by clinical data (efficacy and safety and/or PK/PD) in one therapeutic indication |
| Additional data will be requested in situations like: | |
| 1. Cases in which the biosimilar (the active ingredient of the reference product) interacts with several receptors that can have a different impact in the different proven or unproven therapeutic indications | |
| 2. Situations in which the biosimilar (active ingredient) has more than one active site and the sites can have a different impact in distinct therapeutic indications | |
| 3. Cases in which the therapeutic indication being studied is not relevant for the others in terms of efficacy or safety, that is, lacks sensitivity to detect differences in all the relevant aspects of efficacy and safety | |
| Immunogenicity is related to multiple factors, including the route of administration, dosing regimen, those related to the patient and those related to the disease (for example, concomitant treatments, type of disease, immune status). Thus, immunogenicity can differ among the different therapeutic indications. For this reason, the extrapolation of the immunogenicity of the indication/route of administration being studied for other uses should be justified | |
| FDA24 | If the drug complies with the legal requirements to obtain the license as a biosimilar product under section 351 (k) of the PHS Act on the basis, among other things, of data derived from a clinical study or studies that demonstrate the safety, purity and potency in an appropriate condition of use, the license/authorization can be requested for one or more additional conditions of use (therapeutic indications) for which the reference product is licensed. The scientific justification provided must be sufficient for the extrapolation of clinical data that support the biosimilarity for each condition of use requested |
| Said scientific justification for the extrapolation must include, for example, the following aspects for the proven and extrapolated conditions of use: | |
| • The MOA in each condition of use for which authorization is requested; this can include: | |
| - The antigen/receptor(s) for each relevant activity/function of the product | |
| - Binding, the dose/concentration response and the molecular signaling mechanisms upon association/binding of the receptor(s) and antigen(s) | |
| - Relationship between product structure and target/receptor interactions | |
| - The target/receptor localization and expression | |
| • PK and biodistribution of the product in different patient populations (pertinent pharmacodynamic measures can also provide important information on the MOA) | |
| • Immunogenicity of the product in different patient populations | |
| • The differences in the expected toxicities in each condition of use and in the patient population (including whether the expected toxicities are related to the pharmacological activity of the product or to activities beyond the scope of the target) | |
| • Any other factor that can affect the safety or efficacy of the product in each condition of use and the patient population for which the authorization is requested | |
| The differences between the conditions of use with respect to the factors described above do not necessarily rule out extrapolation. One scientific justification should deal with these differences in the context of the totality of the tests that support a demonstration of biosimilarity | |
| On choosing the condition of use that will enable the subsequent extrapolation of clinical data to other conditions of use, the FDA recommends that the sponsor considers choosing a condition of use that will be sensitive enough to detect clinically significant differences between the 2 products | |
| Authorization can only be obtained for a condition of use that has previously been authorized for the reference product | |
EMA, European Medicines Agency; FDA, United States Food and Drug Administration; MOA, mechanism of action; PD, pharmacodynamics; PHS, Public Health Service; PK, pharmacokinetics.
The costs of treatment go beyond the prices at which it is offered. We know that a drug is competitive not only in efficacy and safety, but also when the price is reasonable and there is an value added in relation to the benefit granted to the patient and to the health institution. The main advantage that should be associated with the use of biosimilars is competitiveness. In Mexico, in January 2017, the cost of the authorized biosimilar Remsima® was $7370/100mg, while the innovator drug, Remicade®, had a cost of $10,405/100mg; that is, if there is an economic benefit in terms of the cost of the drug, nevertheless, the biosimilar still has no program involving support to patients, whereas the innovator does offer an extensive program of support to patients.35
Regulatory FrameworkThe requirements for obtaining approval as biosimilars from the Food and Drug Administration, Health Canada and the EMA include extensive studies both in vitro, demonstrating their similarity to the reference drugs in terms of quality attributes, and preclinical and clinical trials showing their pharmacokinetic comparability, as well as their efficacy, safety and immunogenicity.24,27,33 At the present time, in Mexico, biosimilar drugs must maintain their registration under the tests established in the Mexican official regulations—177 (2013), 257 (2014), 059 (2015), 164 (2015) and 220 (2015). Local laws, standards and guidelines of worldwide reference are in consonance with 4 basic principles proposed by the World Health Organization in matters of biosimilars, which, briefly, establish that: (1) it is necessary to present studies of the physicochemical characterization that show that the heterogeneities encountered in the chemical structure of the biosimilar drug have no negative impact on the efficacy and safety of the treatment; (2) it is necessary to present studies of the biological activity when demonstrated in relevant experimental models showing that the biosimilar has the same mechanism of action as the innovator; (3) it is necessary to present clinical trials involving patients who have the disease in question with a representative sample size in order to demonstrate whether or not the biosimilar is a therapeutic equivalent or not inferior to the innovator drug; and (4) it is necessary to present pharmacovigilance studies that demonstrate that the biosimilar does not represent a greater safety risk than the innovator product.
Systematic Literature ReviewIn order to define the position of the CMR based on the best available evidence, we performed a systematic review of the literature concentrated on clinical trials involving biosimilars that have been proposed for use in rheumatic diseases. We employed the terms “biosimilar”, “biosimilars”, “infliximab”, “adalimumab” and “etanercept” in a search carried out in the PubMed and Cochrane Library databases, utilizing the filter function in the search for randomized clinical trials. We did a manual search to locate studies involving intended copies that were or were not available in Mexico, such as Kikuzubam® and Infinitam® (Kikuzubam® is no longer available in Mexico, although Infinitam® can be purchased). Fig. 1 displays the flow chart corresponding to the search for publications to be considered in this systematic review. The first stage of the analysis consisted in reviewing the title and abstract of the articles encountered; the review was performed in duplicate on the part of 2 rheumatologists who selected the articles that met the selection criteria. Disagreements were resolved by a third person. The articles that were selected on the basis of their title and abstract were subsequently reviewed in full by both reviewers to determine those that met the selection criteria. Once identified, their reference lists were examined to check for other articles that could be of interest. The complete texts of all the articles thus identified were reviewed and those that met the selection criteria were included. The selection criteria were as follows: (1) publications in English or Spanish; (2) clinical trials, extensive studies or Mexican registries; (3) evaluation of efficacy, safety, extension of indications, interchangeability and immunogenicity; (4) drugs employed any of the following diseases: RA, juvenile idiopathic arthritis, spondyloarthritis, ankylosing spondylitis, psoriatic arthritis and systemic lupus erythematosus; (5) inclusion of children, adolescents and/or adults; and (6) published since June 2016. The data were extracted and the results of the review were presented to rheumatologists who were members of the CMR in the second International Symposium of Rheumatoid Arthritis, held from September 29 to October 1 of 2016 in Puerto Vallarta in Mexico. During that meeting, the proposed position was conveyed to the members, was voted on and the comments that arose during the voting process were discussed. The members of the CMR who did not attend the meeting were invited to participate in the voting of the position via internet and were given the opportunity to comment on it. In all, between the attendees and members who voted via Internet, 74 rheumatologists participated.
Results
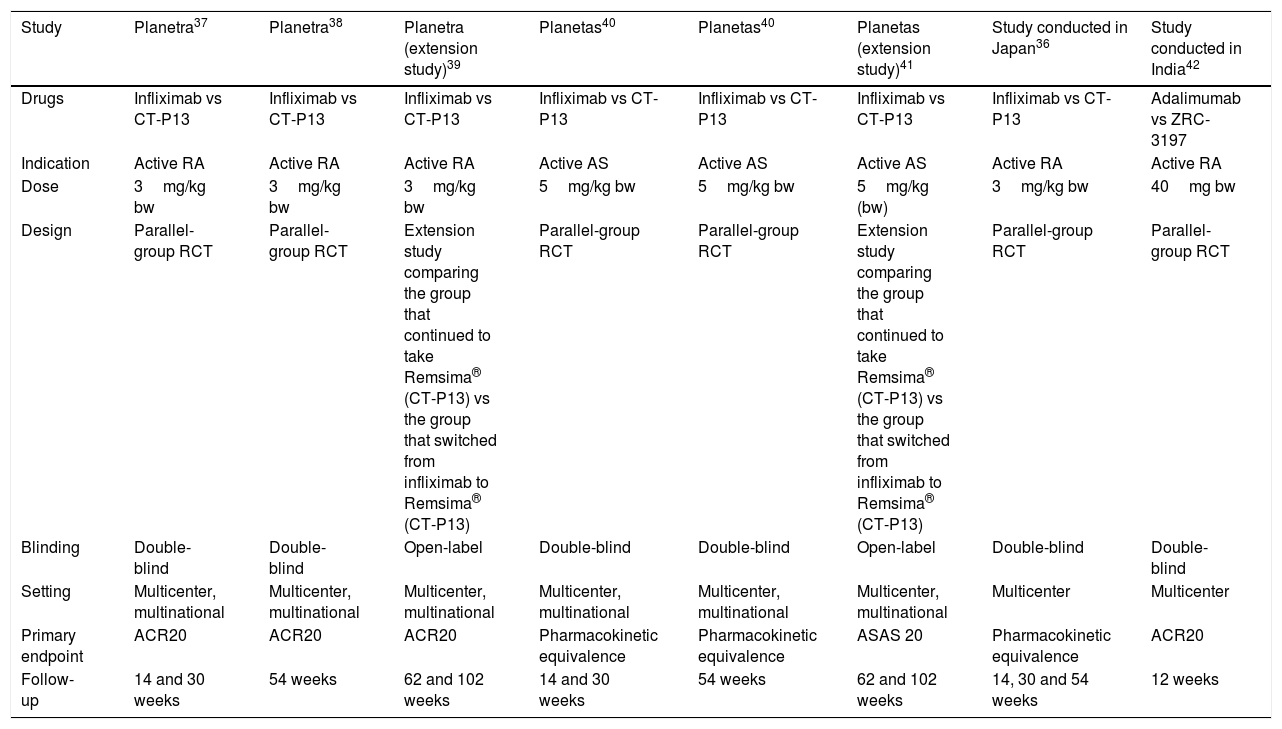
We obtained no results in the search for information on the unclassified biosimilars Kikuzubam® and Infinitam®; there is no available literature on the efficacy and safety of these products. The search for biosimilars in PubMed and Cochrane Library returned 212 articles; once the 85 repeated titles were eliminated, there remained 127 articles for the review of the title and abstract. That left 109 that were not eligible. In 18, the full text was evaluated; of these, only 5 met the inclusion criteria. The review of the references of the related articles led to the identification of 3 more reports. In all, 8 articles were included. The general characteristics of the studies are shown in Table 2. Of the 8 publications included, 7 are on the biosimilarity of infliximab and 1 on the biosimilarity of adalimumab. Of the publications related to the biosimilarity of infliximab, 1 concerns a study performed in Japan,36 3 publications have to do with the PLANETRA multinational study37–39 and 3 with the PLANETAS multinational study.40,41 The publication on the biosimilarity of adalimumab involves a multicenter study conducted in India.42 All of the reports compare the infliximab biosimilar CT-P13 to the original infliximab molecule and all the investigations were financed by Celltrion, the company that developed the biosimilar; comparable results were not obtained with etanercept. The researchers of the PLANETRA and PLANETS studies published their results in week 3037,40; subsequently, they published their findings after 54 weeks of follow-up,38,43 after which they performed an extension study39,41 in which the group of patients receiving the biosimilar drug continued with the same therapy and the group that had been treated at first with the reference product switched to the biosimilar starting in week 62 until week 102.
General Characteristics of the Studies Included in the Systematic Review.
| Study | Planetra37 | Planetra38 | Planetra (extension study)39 | Planetas40 | Planetas40 | Planetas (extension study)41 | Study conducted in Japan36 | Study conducted in India42 |
|---|---|---|---|---|---|---|---|---|
| Drugs | Infliximab vs CT-P13 | Infliximab vs CT-P13 | Infliximab vs CT-P13 | Infliximab vs CT-P13 | Infliximab vs CT-P13 | Infliximab vs CT-P13 | Infliximab vs CT-P13 | Adalimumab vs ZRC-3197 |
| Indication | Active RA | Active RA | Active RA | Active AS | Active AS | Active AS | Active RA | Active RA |
| Dose | 3mg/kg bw | 3mg/kg bw | 3mg/kg bw | 5mg/kg bw | 5mg/kg bw | 5mg/kg (bw) | 3mg/kg bw | 40mg bw |
| Design | Parallel-group RCT | Parallel-group RCT | Extension study comparing the group that continued to take Remsima® (CT-P13) vs the group that switched from infliximab to Remsima® (CT-P13) | Parallel-group RCT | Parallel-group RCT | Extension study comparing the group that continued to take Remsima® (CT-P13) vs the group that switched from infliximab to Remsima® (CT-P13) | Parallel-group RCT | Parallel-group RCT |
| Blinding | Double-blind | Double-blind | Open-label | Double-blind | Double-blind | Open-label | Double-blind | Double-blind |
| Setting | Multicenter, multinational | Multicenter, multinational | Multicenter, multinational | Multicenter, multinational | Multicenter, multinational | Multicenter, multinational | Multicenter | Multicenter |
| Primary endpoint | ACR20 | ACR20 | ACR20 | Pharmacokinetic equivalence | Pharmacokinetic equivalence | ASAS 20 | Pharmacokinetic equivalence | ACR20 |
| Follow-up | 14 and 30 weeks | 54 weeks | 62 and 102 weeks | 14 and 30 weeks | 54 weeks | 62 and 102 weeks | 14, 30 and 54 weeks | 12 weeks |
ACR, American College of Rheumatology; AS, ankylosing spondylitis ASAS, Assessment in Ankylosing Spondylitis; bw, body weight; RA, rheumatoid arthritis; RCT, randomized controlled trial.
The pharmacokinetics and pharmacodynamics of biosimilar CT-P13 were found to be comparable to those of infliximab in all of the studies. The efficacy was measured on the basis of the multiple indicators used in the 7 publications. The PLANETRA study37,38 evaluated the American College of Rheumatology (ACR20, ACR50 and ACR70) response, the Simplified Disease Activity Index (SDAI), good or moderate European League Against Rheumatism (EULAR) response, low activity or remission (defined by the Disease Activity Score 28/C-reactive protein [DAS28-CRP]), the need for rescue therapy and ACR/EULAR remission, and found no significant differences in weeks 14, 30 and 54. Again, the extension study39 encountered no differences in any of the indicators in weeks 62 and 102 when comparing the group that only took CT-P13 and the group that switched from infliximab to CT-P13. The study of the adalimumab biosimilar42 did not assess either pharmacokinetics or pharmacodynamics.
The PLANETAS study40 evaluated, at weeks 14 and 30, the Assessment in Ankylosing Spondylitis (ASAS 20 and ASAS 40) criteria, changes in Ankylosing Spondylitis Disease Activity Score (ASDAS) – CRP, changes in Bath Ankylosing Spondylitis Disease Activity Index (BASDAI), changes in Bath Ankylosing Spondylitis Functional Index (BASFI) and Bath Ankylosing Spondylitis Metrology Index (BASMI); in follow-up week 54,43 it evaluated ASAS 20, ASAS 40, ASAS partial remission, changes in ASDAS-CRP, changes in BASDAI, changes in BASFI, and changes in BASMI. The authors observed no significant differences between the groups taking CT-P13 and infliximab. Again, the expansion study40 showed no differences in any of the indicators at weeks 62 and 102 in the group that took only CT-P13 and the group that switched from infliximab to CT-P13.
The report by Takeuchi et al.36 evaluated ACR20, ACR50, ACR70, the percentage of EULAR responders, changes in DAS28 – erythrocyte sedimentation rate (ESR), changes in DAS28-CRP, changes in SDAI and in Clinical Disease Activity Index (CDAI) at weeks 14, 30 and 54; they only observed differences in the changes in SDAI, which were greater in the CT-P13 group than in the infliximab group (−18.4±15.8 vs −14.1±12.2; P<.05). The article of Jani et al. evaluated ACR20, ACR50 and ACR70, and found no differences between adalimumab and its biosimilar ZRC-3197.42
Safety was evaluated in all of the studies in phases involving parallel groups. The proportion of unfavorable events and adverse reactions that developed in at least 1% of the patients in the CT-P13 group was, respectively, 64.8% and 34.3% in the PLANETRA study, 60.1% and 35.2% in the PLANETAS study and 88.2% and 84.3% in the study by Takeuchi et al. In the infliximab group the rates were 63.9% and 26.3% in the PLANETRA study, 60.8% and 35.9% in the PLANETAS study and 86.8% and 88.1% in the study published by Takeuchi et al. There were no statistically significant differences. In the study by Jani et al., during the 12-week follow-up, 60 participants included in each group developed 13 adverse events in 7 patients in the group treated with biosimilar of adalimumab, whereas in the group receiving the reference product, there were 15 adverse events in 10 patients; these differences were not statistically significant.
The results on safety in the extension studies were as follows: the PLANETAS study reported that the proportion of patients who had at least 1 adverse event was 40.9% in the maintenance group, in contrast to the group that switched from the innovator to CT-P13, in which the proportion was 71.4%; the PLANETRA extension study reported a similar proportion of adverse events (53.5% vs 53.8%).
The PLANETRA study, performed in patients with RA, reported the detection of immunogenicity in the CT-P13 and infliximab groups of 25.4% and 25.8% in week 14, of 48.4% and 48.2% in week 30, and of 49.1% and 48.3%, respectively, in week 54. In the extension study, immunogenicity was detected in 44.7% of the group that continued to take CT-P13 and in 46.2% of the participants who switched to the biosimilar in week 78 and in 40.3% and 44.8%, respectively, in week 102; none of the differences were statistically significant. The article of Takeuchi et al.,36 which also involved RA patients, reported immunogenicity in week 14 in 19.6% of the patients who received CT-P13 and in 15.1% of those treated with infliximab; immunogenicity was detected in 25.5% and 26.4% in week 30 and in 25.5% and 32.1%, respectively, in week 54. None of the differences were statistically significant.
The PLANETAS study, carried out in patients with ankylosing spondylitis, reported immunogenicity in 9.1% of the CT-P13 group and 11% of the infliximab group in week 14, in 27.4% and 22.5% in week 30, and in 19.5% and 23%, respectively, in week 54. In the extension study, immunogenicity was reported in week 78 in 23.3% of the group that continued with CT-P13 and in 29.8% of those who switched and in 23.3% and in 27.4%, respectively, in week 102. None of the differences were statistically significant. The study of Jani et al.42 reported immunogenicity in follow-up week 12 in 2 of the 60 participants taking the biosimilar of adalimumab and in 1 of the 60 in the group being treated with the reference product.
DiscussionGiven the lack of scientific evidence on the efficacy, safety, immunogenicity and interchangeability of still unclassified biological products, such as Kikuzubam® and Infinitam®, it is impossible to organize a discussion concerning their use. Biosimilars may offer a less costly alternative than existing biological products when their patents expire and favor clean competition, thus improving access to biological drugs of a larger number of patients and, therefore, contributing to the financial sustainability of health systems.21 Nevertheless, literature concerning the effect of biosimilar drugs in rheumatic diseases continues to be limited. Although there are more studies on the pharmacokinetics, pharmacodynamics, efficacy, safety and immunogenicity of biosimilars, many of them have been carried out in healthy individuals44–47 or with other types of conditions, such as inflammatory bowel disease or skin diseases.
On the basis of the results of the studies included in the systematic review, we can assert that there is sufficient evidence to accept the pharmacokinetic and pharmacodynamic comparability between CT-P13 and its reference product, infliximab. It has been reported in other sources that CT-P13 is identical to the innovator product in amino acid sequence, pharmaceutical form, potency, composition and route of administration.48 We found only 1 study on a biosimilar of adalimumab that evaluated efficacy, safety and immunogenicity in RA patients, although the need for additional studies on safety and effectiveness will depend on factors such as uncertainty concerning the effect of the drug or the understanding of the mechanisms of action of the product and the pathophysiology of the disease. Achieving the goal of demonstrating that there are no clinically significant differences between the reference product and the biosimilar generally requires at least studies on human pharmacokinetics and evaluations of immunogenicity.23
The results in terms of efficacy of CT-P13 in RA and ankylosing spondylitis are comparable to those obtained with the reference product. Nevertheless, the main objective in the report by Takeuchi et al. and the PLANETAS study was not efficacy, but the pharmacokinetic equivalence; only the PLANETRA study had as a primary endpoint the assessment of the efficacy using the ACR20 response. Despite being the indicator requested by international agencies in order to grant approval for its marketing, it is not the indicator with the greatest clinical relevance. Therefore, it should be considered that, since there are no studies designed to evaluate the clinically significant indicators of efficacy with CT-P13, it cannot be assured that there is sufficient statistical power to observe differences between the groups. The study of Jani et al.42 did have as its primary endpoint the clinical efficacy measured with ACR20 and, thus, there is information that confirms that, in terms of efficacy, there are no differences between the innovator and the biosimilar; however, ACR20 is not necessarily the indicator of the success of the treatment most widely utilized in clinical practice.
None of the studies involving parallel groups showed differences between the group receiving the reference product and that taking the biosimilar; an important difference was observed only in the PLANETAS extension study, since the group that switched from the innovator to the biosimilar had an increase in the development of adverse events of 30% compared with the group that continued with the innovator. The sample size of the studies included in this review makes it impossible to clearly determine the safety profile of the treatment strategies. Pharmacovigilance studies are necessary to provide tools with which to carry out this evaluation.
The results in terms of immunogenicity, there were no significant differences between the groups either in the parallel-group studies or in the extension studies with switching from the innovator product to the biosimilar. Nevertheless, it is surprising to see the high percentage of participants who developed antibodies to these biological drugs. While the “humanization” of murine monoclonal antibodies has greatly improved their in vivo tolerability, some humanized sequence-derived antibodies, even those that are “fully” human, still carry immunogenic potential.49 The high percentage of immunogenicity cannot only be attributed to the reference product or to the biosimilar, and it has been proposed that immunogenicity will always be present in the case of therapeutic use of antibodies due to the nature of the antigen-specific combining sites.49 In the PLANETRA study, 25% of the participants developed immunogenicity by week 14 and nearly half by week 30, in contrast to the study by Takeuchi et al.; in the latter, although participants with RA were also included, the percentages of immunogenicity were much lower, the highest being 32.1%. Even though one of the major differences between the study of Takeuchi et al. and the PLANETRA study is the ethnic group of the patient population, this does not seem to explain the contrast in the results of the studies. Another investigation compared ethnic sensitivity toward monoclonal antibodies in Japanese and non-Japanese individuals and found that the majority of monoclonal antibodies follow the expected behavior regardless of the ethnicity; the only exception seems to be adalimumab, which did show differences related to ethnicity.50 The immunogenic response could thus be determined more by the pathophysiology of the disease. The PLANETAS study, which included participants with ankylosing spondylitis, detected immunogenicity in quite a lower percentage of the patient population, never surpassing 30%. Other studies have reported reduced immunogenicity among patients with ankylosing spondylitis in comparison with individuals with RA.51 The latter finding brings us to question indication extrapolation, given the differences observed between patients with RA and those with ankylosing spondylitis and the lack of evidence of the efficacy and safety in other indications. It is necessary to observe certain caution when managing these data and utilizing them comparatively and complementarily, since it is necessary to take into account the variability of the results depending on the diagnostic technique utilized. That is, if a potential change in immunogenicity after 12 months of treatment was the major incident to take place, to determine its clinical relevance would require access to a central laboratory as well as correlation of the results with the levels of the drug in the organism, the response to treatment and the incidence and severity of adverse events.21
The data provided during the extension phase to week 102 in the PLANETAS and PLANETRA studies shows that switching the reference product infliximab to its biosimilar CT-P13 has no deleterious effects on the efficacy, safety or immunogenicity compared to the continuous use of CT-P13.52 Moreover, a review of the literature that, in addition to taking into account the abovementioned studies, included real-world data from patients with inflammatory bowel disease, concluded that switching from the reference product to its biosimilar CT-P13 does not have undesirable effects.52 Nevertheless, although the evidence concerning the effect of switching from the reference product to the biosimilar is certainly useful, these data do not respond to the question as to the effect of interchangeability on efficacy, safety and immunogenicity. None of these studies included switching back again to the reference product, a condition that is necessary to evaluate interchangeability.23 It must be remembered that, to show that the substitution of the treatment is feasible, interchangeability must first be demonstrated. As the literature does not include studies on the biosimilar of adalimumab that assess the effect of the change in therapy or interchangeability, it cannot be established whether it is safe to alternate the innovator product and its biosimilar.
PositionThe CMR proposes adopting the following position regarding biosimilar products in Mexico:
- 1.
An innovator biological product is that which has been developed and registered on a worldwide basis for the first time for one or more indications and that a biosimilar is that which, utilizing the same molecular biology techniques, intends to achieve a structure and function comparable to those of the innovator product.
- 2.
The achievement of biosimilar products requires a complex process that does not guarantee that the biosimilar drugs that do not meet the Mexican and international regulatory requirements be identical to the innovators. For this reason, and given the lack of scientific evidence on their efficacy, safety and interchangeability, biosimilars of this type should not be considered interchangeable.
- 3.
The following should be taken into account: (1) in rheumatology, clinical trials with biosimilars have involved patients who had never received biological therapy; (2) there is no evidence of the safety and efficacy of their interchangeability in patients who remain stable with the reference product; and (3) that the information on patient safety during long-term follow-up cannot be adequately assessed if the innovator drug is replaced by a biosimilar and vice versa. It is erroneous to accept the practice of automatic substitution of a drug.
- 4.
Each product should clearly emphasize in its nomenclature whether it is an innovator drug or a biosimilar, and the batch number should be given for the purpose of ensuring the pharmacovigilance of all medications and guarantee the safety of each drug administered to the patient.
- 5.
Those involved in the manufacture, prescription and use of biosimilar products are obliged to exercise long-term pharmacovigilance and that secondary events should be reported to the COFEPRIS web page (www.cofepris.gob.mx).
- 6.
A patient who remains stable with the reference product should not be switched to a biosimilar for economic reasons.
- 7.
In patients who are naïve to biological treatment, the decision on therapy involving approved biological products (reference or biosimilars) should be based on an individual evaluation of the risk/benefit rather than only on economic aspects.
- 8.
Indication extrapolation should not be performed or approved based only on the approval obtained from COFEPRIS for the reference product and in the absence of data on the safety and efficacy of the biosimilar.
- 9.
It is recommended that COFEPRIS, the General Health Council, health institutions, economic institutions and key actors get together to establish a dialog to officially determine the standardization of pricing criteria, interchangeability and substitution of the treatment with the biosimilar drug, with the best available scientific evidence and in benefit of the patients, thus finding a way to comply with the right to health protection specified in the Mexican constitution.
- 10.
This position must be updated every 2 years or whenever important evidence is made known in this regard.
The CMR manifests that this working group is in favor of the development of biosimilar drugs, both in Mexico and in other parts of the world, as well as their approval, without external interference, on the part of regulatory agencies. Aside from demonstrating their pharmacokinetic equivalence, they should be subjected to the highest quality standards in terms of production and development. Their efficacy and safety should be compared with those of the reference product in phase III and IV studies with an adequate statistical power, and must be followed according to a strict pharmacovigilance program. The goals for biosimilar development should include: (1) a substantial savings for public health institutions; (2) the possibility that patients can acquire these drugs as out-of-pocket expenses; (3) access to these drugs of larger sectors of the population; and (4) the condition that the economic factor not be put before therapeutic efficacy and optimal patient safety.
NoteThe opinions expressed in this article are those of the authors and only reflect the position of the CMR. They do not manifest official policy or the position of any of the institutions with which they are associated.
Ethical DisclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that no patient data appear in this article.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflicts of InterestNone of the authors received payment for participation in the present article. Daniel Xibille worked as a consultant and received fees for presentations from Hospira-Pfizer, Eli Lilly, Pfizer, Glaxo Smith Kline, Janssen, Bristol and Abbvie. Sandra Carrillo and Leonardo Limón worked as consultants and received fees for presentations from Eli Lilly, Pfizer, Roche, Janssen, Bristol, Abbvie, Glaxo Smith Kline and Novartis. Abdieel Esquivel has received funds for consultancies, brand counseling, academic events and conferences in Mexico and international meetings from Pfizer, Novartis, Eli Lilly, Abbvie, Sanofi, Hospira, Roche, Janssen, Stendhal, Merck, UCB Pharma and MSD. Gabriela Huerta-Sil, Ramiro Hernández, Guadalupe Olvera-Soto, Luis Javier Jara-Quezada and Marcela Pérez-Rodríguez declare they have no conflicts of interest.
We wish to thank the physicians who participated in the session held to draft the position: Dr. Francisco Javier Aceves Ávila, Dr. Jorge Enrique Aguilar Arreola, Dr. Guadalupe Cecilia Aguilar Domínguez, Dr. Lilia Andrade Ortega, Dr. Ana Cecilia Arana Guajardo, Dr. María de Jesús Araujo Arias, Dr. Marcos Daniel Ayala Arzola, Dr. Jorge Alberto Barragán Garfías, Dr. Mario Alfredo Chávez López, Dr. Lucia Comellas Kirkerup, Dr. José Arturo Covarrubias Cobos, Dr. Beatriz Alicia de Cortina Camou, Dr. Juan Carlos de la Cruz Castillejos, Dr. Karla Irene de la Cruz Rodríguez, Dr. Adrián Alejandro de la Madrid Cernas, Dr. José Noel Díaz Muñoz, Dr. Sandra Enciso Pelaéz, Dr. María Amparo Enríquez Maldonado, Dr. Grissel Espericueta Arriola, Dr. Diana Laura Ferrusquia Toriz, Dr. Sonia de la Mercedes Gacitúa Zambrano, Dr. Víctor Adán Gallaga Gutiérrez, Dr. Oscar Gallegos Hernández, Dr. José Luis García Figueroa, Dr. Conrado García García, Dr. César Leonardo García López, Dr. Imelda García Olivera, Dr. Karla Maritza García Osuna, Dr. Rafael García Rascón, Dr. Claudia Elizabeth Gómez López, Dr. Osvaldo González La Riviere, Dr. Mariana Paola González Mora, Dr. Leticia Gutiérrez Pérez, Dr. Jorge Carlos Guzmán Murillo, Dr. Jaime Hadid Smeke, Dr. Éufrates Hernández Núñez, Dr. María del Carmen Hernández Quiroz, Dr. Guillermo Fernando Huerta Yáñez, Dr. Graciela Ibáñez Landín, Dr. Jorge Jaimes Hernández, Dr. Xochitl Jiménez Jiménez, Dr. Bernardo Julián Martínez, Dr. Montserrat Lamuño Encorrada, Dr. Marysol Lendechy Velázquez, Dr. Luz Aurora León Legaría, Dr. Ana Laura Marines Castillo, Dr. Gloria Esther Martínez Bonilla, Dr. Lucia Verónica Maya Piña, Dr. Claudia Irene Melendez Mercado, Dr. Josefina Xóchitl Mendoza Vázquez, Dr. Juan Manuel Miranda Limón, Dr. Mauricio Montero Luna, Dr. Carlos Moya McClaugherty, Dr. Gracida Karol Mugica de la Lanza, Dr. Roberto Negrete López, Dr. Victoria Obiala Ezenwa, Dr. Francisco Olan, Dr. Liliana Ortiz Martínez, Dr. Leslie Osuna Garibaldi, Dr. Ana Virginia Perla Navarro, Dr. Rebeca Ramírez González, Dr. Greta Cristina Reyes Cordero, Dr. Jacqueline Reyes Rueda, Dr. Humberto Alfredo Ricardez Puente, Dr. Tatiana Sofía Rodríguez Reyna, Dr. Roxana Minerva Rodríguez Romo, Dr. Mauricio Eduardo Rubio Sánchez, Dr. Raymundo Ruíz García, Dr. Miguel Ángel Saavedra Salinas, Dr. Arturo Sánchez Arriaga, Dr. Antonio Sánchez González, Dr. Tania Sánchez Hernández, Dr. Maria del Lucero Sanchez Segura, Dr. Karina Santana de Anda, Dr. Clara Shumski Flaschner, Dr. Sandra Araceli Sicsik Ayala, Dr. Luis Humberto Silveira Torre, Dr. Claudia Magdalena Solís Alvarado, Dr. Leobardo Terán Estrada, Dr. Norma Edith Torres Gudiño, Dr. Ignacio Alfredo Valerio Morales, Dr. Lorenzo Valle Gómez, Dr. Julio Villagómez Calderón, Dr. Arturo Villarreal Ortega, Dr. Erick Adrian Zamora Tehozol and the rest of our colleagues who voted via internet.
Please cite this article as: Xibille D, Carrillo S, Huerta-Sil G, Hernández R, Limón L, Olvera-Soto G, et al. Escenario actual de los medicamentos biocomparables en México: posicionamiento del Colegio Mexicano de Reumatología, 2016. Reumatol Clin. 2018;14:127–136.


