

ILD patients can be positive to highly specific autoantibodies of connective tissue diseases (CTD). Among them stand out myositis-specific and associated autoantibodies (MSA/MAA). There is limited knowledge about treatment response and prognosis of ILD patients positive to MSA/MAA (MSA/MAA-ILD). Our aim was to describe clinical, radiological and pulmonary function (PF) of MSA/MAA-ILD Latin-American patients and risk factors associated to PF at onset and long term follow up.
MethodsMulticentric retrospective study of MSA/MAA-ILD patients evaluated between 2016 and 2018 in 3 ILD clinics in Latin America. Clinical, functional and tomographic variables were described. Variables associated with poor baseline PF and associated with functional improvement (FI) were analyzed in a multivariate logistic regression model.
ResultsWe included 211 patients, 77.4% female, mean age 57 years old. Most frequent MSA/MAA were Ro-52 and Jo-1. Poor baseline PF was associated to ILD as initial diagnosis and NSIP/OP HRCT pattern. 121 patients were included in the follow up PF analysis: 48.8% remained stable and 33% had a significant FI. In multivariate analysis, OP pattern on HRCT was associated with FI. Systemic symptoms from the beginning and the absence of sclerodactyly showed a trend to be associated with FI.
ConclusionsWorse baseline PF could be related to the absence of extra-thoracic symptoms and “classic” antibodies in CTD (ANA), which causes delay in diagnosis and treatment. In contrast, FI could be related to the presence of extra-thoracic signs that allow timely diagnosis and therapy, and more acute and subacute forms of ILD, such as OP pattern.
Los pacientes con enfermedad pulmonar intersticial (EPI) pueden presentar positividad para autoanticuerpos específicos de enfermedades autoinmunes, como los anticuerpos mioespecíficos (AME) o mioasociados (AMA). Existe escasa información disponible sobre pronóstico y respuesta al tratamiento de estos pacientes. Nuestro objetivo fue describir las características clínicas, radiológicas, funcionales y pronósticas de una cohorte latinoamericana de pacientes con EPI y AME/AMA.
MétodosEstudio retrospectivo multicéntrico de una cohorte pacientes con EPI y AME/AMA, evaluados en tres centros latinoamericanos entre 2016 y 2018. Describimos variables clínicas, tomográficas y funcionales. Analizamos variables asociadas con valores funcionales bajos al inicio y mejoría funcional mediante regresión logística.
ResultadosIncluimos 211 pacientes, 77,4% mujeres, con una media de edad de 57 años. Los anticuerpos más frecuentes fueron Ro-52 y Jo-1. Valores funcionales bajos al inicio se asociaron con la presencia de EPI desde el debut y con un patrón de neumonía intersticial no específica (NINE)/neumonía organizativa (NO) en la tomografía computarizada de alta resolución (TACAR). Se analizó la evolución funcional en 121 pacientes: 48% permanecieron estables y 33% presentaron mejoría. El patrón de NO en TACAR, se asoció significativamente con mejoría funcional, mientras que la presencia de EPI desde el debut y la ausencia de esclerodactilia mostraron una tendencia en el análisis multivariado.
ConclusionesValores funcionales bajos al debut podrían asociarse con la ausencia de síntomas extratorácicos al inicio, por llevar a un diagnóstico y tratamiento tardíos. Por el contrario, la presencia de síntomas sistémicos al debut, junto con formas más agudas de EPI como NINE/NO podrían asociarse con mejoría funcional por permitir un diagnóstico y tratamiento precoz.
Interstitial lung disease (ILD) comprise a heterogeneous group of pulmonary diseases that affect the lung interstitium and can present with different radiological and histopathological patterns. Main causes of ILD are idiopathic pulmonary fibrosis (IPF), environmental exposures and connective tissue diseases (CTD).1 An important proportion of patients with ILD are positive to highly specific autoantibodies, but many of these patients may not be classified as specific CTD according current classification criteria. For example, those with positive myositis-specific autoantibodies (MSA) such as the anti-aminoacyl tRNA synthetases (anti tRNAs: anti Jo-1, anti PL-7, anti PL-12, anti Ej, anti Oj, anti KS, anti Zo, anti Ha), and anti MDA-5 or myositis-associated antibodies (MAA) like anti PM/SCL, anti Ku, or anti Ro-52, among others.2
The myositis or idiopathic inflammatory myopathies (IIM) are a group of autoimmune diseases that share some clinical manifestations such as muscle inflammation, skin rash, arthritis, and high prevalence of ILD. Different phenotypes among the IIM are closely related to the subtype of MSA and MAA,2 and although it is well recognized that ILD is a very important manifestation of some IIM in the context of MSA/MAA, ILD is not included in the EULAR/ACR classification criteria for IIM.3 So, a great proportion of ILD patients positive to MSA/MAA may not be classified as IIM, but may be classified as interstitial pneumonia with autoimmune features (IPAF) according to the proposal of the American Thoracic Society/European Respiratory Society (ATS/ERS).4
At present, there is limited knowledge about the differences in clinical manifestations, response to treatment, pulmonary function and prognosis of ILD patients positive to MSA/MAA. These are diverse autoantibodies, and as we mentioned before, they are associated to different phenotypes and perhaps different prognosis.
With this background, we designed this study with the aim to describe the clinical, radiological and pulmonary function of Latin-American patients with ILD positive to MSA/MAA and evaluate risk factors associated to pulmonary function at onset and long term follow up.
MethodsWe conducted a multicentric retrospective cohort study. We included patients with ILD and MSA/MAA evaluated between 2016 and 2018 in 3 multidisciplinary ILD clinics in Argentina (Hospital María Ferrer), Chile (Instituto Nacional del Tórax) and Mexico (Instituto Nacional de Enfermedades Respiratorias). Every patient was confirmed to have ILD by thoracic high-resolution computed tomography (HRCT) and their medical records were reviewed. All patients were evaluated by a multidisciplinary team of pulmonologists, immunologists/rheumatologists and radiologists.
The following variables were described: age, gender, tobacco exposure, gastroesophageal reflux disease (GERD) symptoms, dyspnea severity (assessed with the mMRC scale) and treatment schemes. We registered the following data related with the presence of an underlying CTD: arthritis, muscle weakness, mechanic's hands (distal digital hyperkeratosis and fissuring), fever, Raynaud's phenomenon, sclerodactyly, puffy hands, telangiectasias, skin ulcers, sicca symptoms (xerostomia and/or xerophthalmia) and dermatomyositis rash (heliotrope rash, Gottron's papules, shawl sign). These were called systemic symptoms.
Pulmonary function testsWe evaluated the pulmonary functional status using the percentage of predicted forced vital capacity (FVC%) and the percentage of predicted carbon monoxide diffusing capacity (DLCO%) at the time of diagnosis and every 4–6 months according to reference procedures guidelines. Pulmonary functional tests were performed according to international standard.5–7 Clinical and serological variables associated with poor baseline pulmonary function and variables associated with longitudinal pulmonary function were analyzed. Poor baseline lung function was defined as FVC <70% of predicted and/or DLCO <60% of predicted.
To identify factors associated with functional improvement, patients with less than 3 months of follow up were excluded. Functional improvement was considered an increase in the percentage FVC greater than 10%. Improvement in FVC was calculated by considering the difference between percentages of predicted values.
HRCTsHRCTs were evaluated by ILD expert radiologists that were blinded to clinical data. HRCT patterns were classified as usual interstitial pneumonia (UIP), nonspecific interstitial pneumonia (NSIP), organizing pneumonia (OP), or an overlap of both (NSIP/OP) based on the 2013 Idiopathic Interstitial Pneumonia guidelines.1
Laboratory tests and autoantibodiesBaseline laboratory values including serum creatin kinase levels were obtained. Patients were evaluated with an autoimmune panel searching for autoantibodies such as: antinuclear antibodies (ANA), anti-citrullinated cyclic peptide (CCP), anti RNP, anti SM, anti Ro/SS-A, anti La/SS-B, anti Ro-60, anti Ro-52, anti SCL-70, anti Jo-1, anti EJ, anti OJ, anti PL-7, anti PL-12, anti PM/SCL-75, anti PM/SCL-100, anti Ku, anti Mi2, anti SRP, anti NPX2, anti TIF1γ, anti SAE, anti MDA-5. ANAs were measured by indirect immunofluorescence (IFI). MSA/MAA determination was performed by immunoblot (Autoimmune Inflammatory Myopathies 16 Ag, Euroline®, Euroimmun, Lübeck, Germany in Chile and Mexico; and Myositis LIA PL®, Imtec-HUMAN, Germany in Argentina).
Statistical analysisQualitative variables were analyzed by Chi-square test or Fischer's exact test. Continuous variables were expressed as mean with standard deviation (SD) or median with inter-quartile range (IQR).
Variables associated with functional improvement were analyzed in a univariate logistic regression model. Variables with p values of 0.1 or less were included in the multivariate analysis. The decision to include a variable in the definitive model was guided with the likelihood ratio test. The discriminative capacity of the model was expressed as area under the receiver operating curve (AUC). The Hosmer–Lemeshow goodness of fit test was used to evaluate the model calibration. The manuscript was written according to STROBE initiative for the communication of observational studies.8
Ethical issuesThe study was submitted and approved by local internal review boards in each center.
ResultsWe included 211 patients. Chile 119 (56.4%), Mexico 50 (23.7%) and Argentina 42 (19.9%). The majority of patients were women (77.4%), with a mean age of 57 years (SD: 12), and 34% were past or current smokers (Table 1).
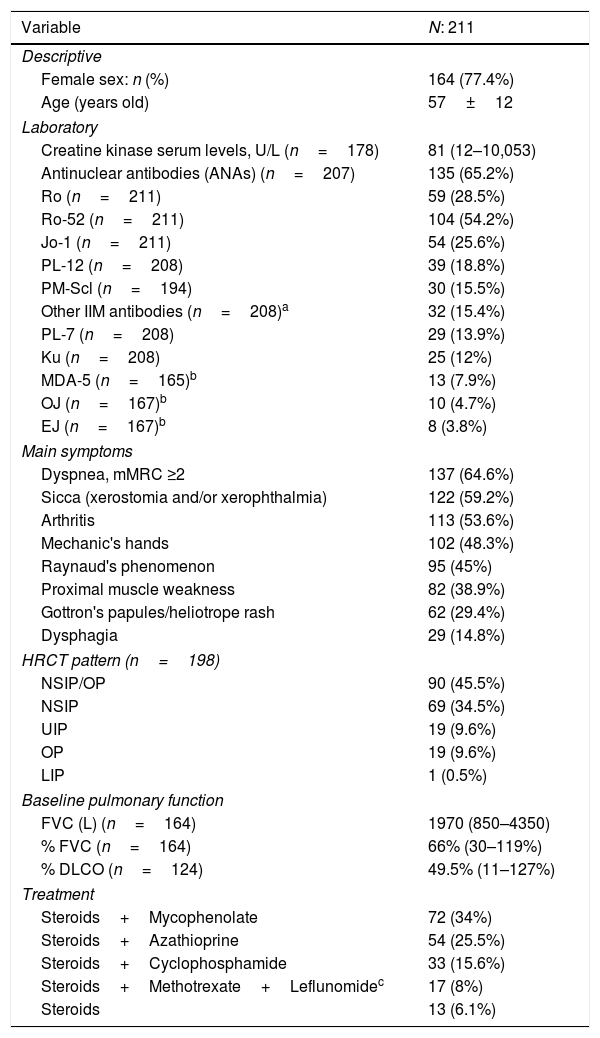
Description of demographic, clinical and tomographic variables.
| Variable | N: 211 |
|---|---|
| Descriptive | |
| Female sex: n (%) | 164 (77.4%) |
| Age (years old) | 57±12 |
| Laboratory | |
| Creatine kinase serum levels, U/L (n=178) | 81 (12–10,053) |
| Antinuclear antibodies (ANAs) (n=207) | 135 (65.2%) |
| Ro (n=211) | 59 (28.5%) |
| Ro-52 (n=211) | 104 (54.2%) |
| Jo-1 (n=211) | 54 (25.6%) |
| PL-12 (n=208) | 39 (18.8%) |
| PM-Scl (n=194) | 30 (15.5%) |
| Other IIM antibodies (n=208)a | 32 (15.4%) |
| PL-7 (n=208) | 29 (13.9%) |
| Ku (n=208) | 25 (12%) |
| MDA-5 (n=165)b | 13 (7.9%) |
| OJ (n=167)b | 10 (4.7%) |
| EJ (n=167)b | 8 (3.8%) |
| Main symptoms | |
| Dyspnea, mMRC ≥2 | 137 (64.6%) |
| Sicca (xerostomia and/or xerophthalmia) | 122 (59.2%) |
| Arthritis | 113 (53.6%) |
| Mechanic's hands | 102 (48.3%) |
| Raynaud's phenomenon | 95 (45%) |
| Proximal muscle weakness | 82 (38.9%) |
| Gottron's papules/heliotrope rash | 62 (29.4%) |
| Dysphagia | 29 (14.8%) |
| HRCT pattern (n=198) | |
| NSIP/OP | 90 (45.5%) |
| NSIP | 69 (34.5%) |
| UIP | 19 (9.6%) |
| OP | 19 (9.6%) |
| LIP | 1 (0.5%) |
| Baseline pulmonary function | |
| FVC (L) (n=164) | 1970 (850–4350) |
| % FVC (n=164) | 66% (30–119%) |
| % DLCO (n=124) | 49.5% (11–127%) |
| Treatment | |
| Steroids+Mycophenolate | 72 (34%) |
| Steroids+Azathioprine | 54 (25.5%) |
| Steroids+Cyclophosphamide | 33 (15.6%) |
| Steroids+Methotrexate+Leflunomidec | 17 (8%) |
| Steroids | 13 (6.1%) |
In 146 patients (70%) ILD was diagnosed first, in 43 patients (18%) ILD and CTD were diagnosed simultaneously and CTD was diagnosed first in 25 patients (12%). The mean interval between ILD and CTD diagnosis was 7.6 months.
Patients with positive ANA were 135 (65.2%), with speckled and homogeneous patterns being the most prevalent (22.1% and 17.9%, respectively). The most frequent MSA/MAA were: Ro-52 (54.2%), Jo-1 (25%), PL-12 (18.8%), PM-Scl 75-100 (15.5%), PL-7 (13.9%) and Ku (12%) (Table 1).
The most frequent extra-thoracic symptoms were sicca symptoms (59.2%), arthritis (53.6%), mechanic's hands (48.3%) and Raynaud's (45%). Less than 50% had muscle weakness (Table 1). Almost one third of patients (27.1%) had diagnosis of a CTD different from IIMs, mainly Sjögren's syndrome and systemic sclerosis (SSc). The most frequent radiological patterns were: NSIP/OP (45.5%) and NSIP (34.5%). UIP pattern was found in only 9.6% of patients (Table 1).
Baseline lung functionMedian baseline FVC was 66% of predicted value (IQR 30–119%) and median DLCO was 49.5% of predicted value (IQR 11–127%) (Table 2).
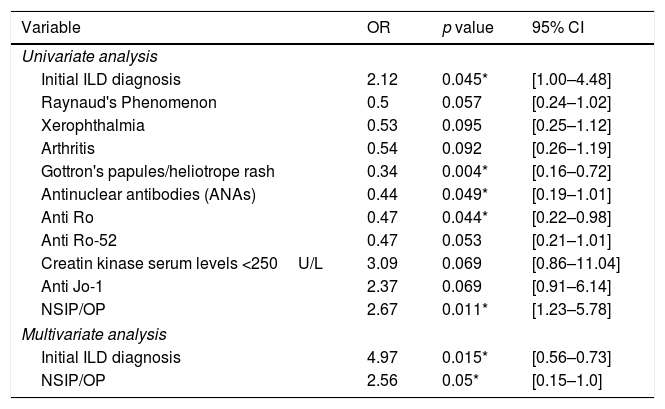
Uni and multivariate analysis for variables associated with poor baseline pulmonary function.
| Variable | OR | p value | 95% CI |
|---|---|---|---|
| Univariate analysis | |||
| Initial ILD diagnosis | 2.12 | 0.045* | [1.00–4.48] |
| Raynaud's Phenomenon | 0.5 | 0.057 | [0.24–1.02] |
| Xerophthalmia | 0.53 | 0.095 | [0.25–1.12] |
| Arthritis | 0.54 | 0.092 | [0.26–1.19] |
| Gottron's papules/heliotrope rash | 0.34 | 0.004* | [0.16–0.72] |
| Antinuclear antibodies (ANAs) | 0.44 | 0.049* | [0.19–1.01] |
| Anti Ro | 0.47 | 0.044* | [0.22–0.98] |
| Anti Ro-52 | 0.47 | 0.053 | [0.21–1.01] |
| Creatin kinase serum levels <250U/L | 3.09 | 0.069 | [0.86–11.04] |
| Anti Jo-1 | 2.37 | 0.069 | [0.91–6.14] |
| NSIP/OP | 2.67 | 0.011* | [1.23–5.78] |
| Multivariate analysis | |||
| Initial ILD diagnosis | 4.97 | 0.015* | [0.56–0.73] |
| NSIP/OP | 2.56 | 0.05* | [0.15–1.0] |
Poor baseline lung function was associated to the presence of ILD at diagnosis of CTD (OR 4.97; p=0.015) and NSIP/OP pattern on HRCT (OR 2.56; p=0.05). Other manifestations such as Gottron's papules, heliotrope rash, positive ANA and anti-Ro antibodies were protective factors and were associated with better lung function at baseline in the univariate analysis (OR 0.34; p=0.004, OR 0.44; p=0.049 and OR 0.47; p=0.044), respectively. In the multivariate analysis these results were not significant (Table 2).
All patients received corticosteroids and main first line immunosuppressants were mycophenolate mofetil (34%, n=72), azathioprine (25.5%, n=54) and cyclophosphamide (15.6%, n=33), similar in all three countries (Table 1).
Variables associated with functional improvementA total of 121 (57%) patients were included in pulmonary function follow-up analysis. Of them, 59 remained stable (48.76%), 40 (33.06%) showed a significant FVC improvement and 22 (18.18%) had a significant decline. The median of functional follow-up was 19.5 (IQR 9–40) months. Only three patients (2.5%) died during the follow up.
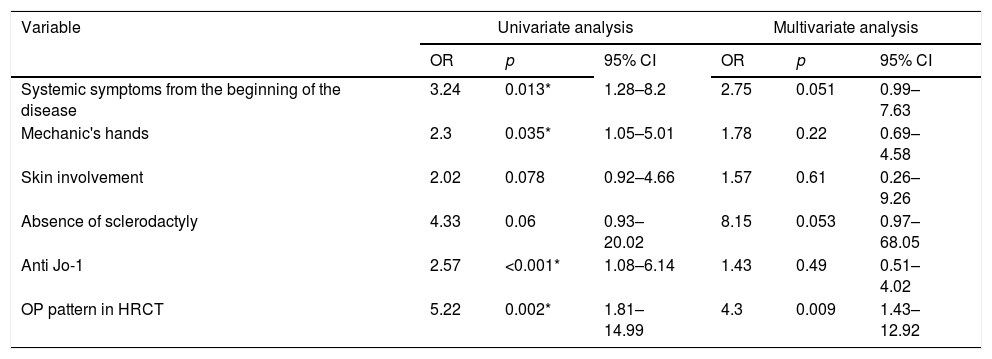
Variables that showed association with functional improvement in the univariate analysis were systemic symptoms from the beginning of the disease, three variables related with cutaneous compromise (mechanic's hands, skin involvement and absence of sclerodactyly), Jo-1 antibody and OP pattern on HRCT (Table 3).
Univariate and multivariate analysis of variables associated with functional improvement.
| Variable | Univariate analysis | Multivariate analysis | ||||
|---|---|---|---|---|---|---|
| OR | p | 95% CI | OR | p | 95% CI | |
| Systemic symptoms from the beginning of the disease | 3.24 | 0.013* | 1.28–8.2 | 2.75 | 0.051 | 0.99–7.63 |
| Mechanic's hands | 2.3 | 0.035* | 1.05–5.01 | 1.78 | 0.22 | 0.69–4.58 |
| Skin involvement | 2.02 | 0.078 | 0.92–4.66 | 1.57 | 0.61 | 0.26–9.26 |
| Absence of sclerodactyly | 4.33 | 0.06 | 0.93–20.02 | 8.15 | 0.053 | 0.97–68.05 |
| Anti Jo-1 | 2.57 | <0.001* | 1.08–6.14 | 1.43 | 0.49 | 0.51–4.02 |
| OP pattern in HRCT | 5.22 | 0.002* | 1.81–14.99 | 4.3 | 0.009 | 1.43–12.92 |
In the multivariate analysis, OP pattern on HRCT (OR 4.30; p=0.009) was significantly associated with functional improvement (Table 3). Systemic symptoms from the beginning of the disease (OR 2.75; p=0.051) and the absence of sclerodactyly (OR 8.15; p=0.053) were associated with functional improvement in the evolution with a trend toward statistical significance. Nor the positivity of anti Jo-1, mechanic's hands or skin involvement were associated with improvement in the multivariate analysis. The AUC of the model was 0.73. The calibration accuracy of the final model showed good results (Hosmer–Lemeshow goodness of fit test p=0.96).
DiscussionWe describe the clinical characteristics and variables associated with functional improvement in a cohort of patients with interstitial lung disease and MSA/MAA from three centers specialized in ILD from Latin America.
In our cohort we observed a greater proportion of middle-aged female patients, concordant with previously published literature. Among MSA, anti tRNAS were the most frequently identified, especially anti Jo-1. These autoantibodies are associated with variable presence of ILD, myositis, arthritis, Raynaud's phenomenon, mechanic's hands and fever, configuring the so-called “Anti-synthetase syndrome”, despite the fact that this clinical syndrome has not validated classification criteria yet.9
The Jo-1 antibody is the most frequently identified in our cohort and in other published data. Unlike the rest of the anti tRNAS, it is more often associated with concomitant myositis at debut of the disease. This is important because although the anti tRNAS are closely associated with the spectrum of the IIM, the presence of muscle involvement is not universal. Anti PL-12, PL-7, KS, OJ and, in smaller proportion, anti EJ are associated with isolated ILD at the beginning of the disease and do not usually develop muscle involvement during follow-up.10
The most frequent extrapulmonary manifestations were sicca symptoms, arthritis, mechanic's hands and Raynaud's phenomenon; whereas muscle weakness and elevated muscle enzymes were not a constant finding, similar to those described in other cohorts.11,12
The pattern of NSIP isolated or associated with OP was the most frequently HRCT pattern encountered, similar to the study published by Waseda et al.,13 where the majority of patients with ILD associated with anti tRNAS had an inconsistent pattern with UIP, corresponding to the NSIP and OP patterns. This tomographic pattern is typically described in patients with MSA/MAA, whereas UIP pattern is less frequent.14 ANA was negative in almost a third of the cohort. In the scenario of MSA/MAA, particularly anti tRNAS, this finding is not unusual due to the cytoplasmic localization of the autoantigens. This is important because many patients could erroneously be classified as having an idiopathic interstitial disease if ANA is negative and no extrapulmonary manifestation is present at the time of the evaluation, delaying the correct diagnosis and an adequate treatment.14,15 In the case of our cohort, 70% of the patients received the initial diagnosis of isolated ILD, with a time interval until the final diagnosis of 7.6 months with a maximum interval of 280 months (23 years).
Regarding MAA, anti Ro-52 was identified in almost half of the cohort. This association is described in the literature not only for the spectrum of IIM but also has been identified in other CTDs like SSc and systemic lupus erythematosus. The presence of this autoantibody has been linked to ILD, Raynaud's phenomenon, mechanic's hands and Jo-1. The relationship of anti Ro-52 with prognosis and course of the disease is controversial.16,17 In our cohort the positivity of PM/SCL was identified in 15% of patients. This antibody is mainly found in the superposition of systemic sclerosis with myositis, and is associated with a higher frequency of sclerodactyly, Raynaud's phenomenon and skin manifestations associated with dermatomyositis. It has been suggested that anti tRNAS and PM/SCL antibodies could present a similar spectrum of clinical manifestations and share certain human leukocyte antigens (HLA).12,18 Finally, anti MDA-5 was observed in 8% of the patients in our cohort. This autoantibody test is not universally available in all our ILD clinics, so we do not know if its frequency could be higher in our MSA/MAA-ILD patients. MDA-5 is described to be associated with clinically amyopathic dermatomyositis, digital ulcers and rapidly progressive ILD with poor response to immunosuppressive treatment.19
Only a small percentage of patients received treatment with corticosteroids (CS) as monotherapy; in the majority of cases CS were used in association with mycophenolate, azathioprine or cyclophosphamide. At the moment, no randomized clinical trials have been published to evaluate the effectiveness of these drugs in the treatment of ILD associated with the presence MSA/MAA, and most of the information comes from observational studies.20–22 Therefore, the indication and choice of the immunosuppressive treatment is based on the available information, expert opinion, local experience with a particular drug, tolerance profile and access to certain drugs. Despite differences in treatment strategies among the three centers, most of the patients remained stable or improved after immunosuppressive therapy.
We observed that OP pattern was associated with functional improvement during follow-up, like other studies. This pattern is related to the presence of inflammation in the lung parenchyma and is described within the classification of the idiopathic ILD as reversible, with good response to treatment and a subacute-acute course.23–25 Although not reaching statically significance, the absence of sclerodactyly was a factor associated with FVC improvement. This could be explained because there is a subgroup of patients with MSA/MAA and SSc features overlap.26 In SSc one of the main pathogenic mechanism is vascular damage involving microvascular endothelial cells leading to fibroproliferative vasculopathy and capillary rarefication and the activation of fibroblasts to myofibroblasts leading to excessive extracellular matrix deposition in skin and blood vessels. It has been described that patients with loss of nailfold capillary density and progressive skin fibrosis within one year of the diagnosis had a FVC decline ≥10% and worse survival during follow up.27–29
The presence of systemic manifestations along with ILD was also related to functional improvement. This could be due to an early diagnosis of the disease and timely treatment, unlike when ILD is the only dominant manifestation.11,12,30 The latter association is in accordance with the fact that Jo-1 positivity was associated with functional improvement in the univariate analysis, as it is described by other authors; but the influence of Jo-1 disappeared in the multivariate analysis. One of the reasons might be that systemic manifestations are more common in anti Jo-1 patients.
Our study has some limitations. First, the inherent limitations of a retrospective study, such as the possible presence of a selection bias. Second, as it is a multicentric cohort study, patients could have been evaluated differently, not all the aforementioned antibodies were evaluated in all patients due to the access of each center to the determinations, so the prevalence of the antibodies may vary. Third, not all patients had baseline pulmonary function (maximum 164/211) or follow-up pulmonary function (maximum 121/211). Finally, the selection criteria for immunosuppressive treatment may have varied according to the preferences of each center and/or access to different drugs.
Despite these aspects, this study emphasizes the importance of searching for signs and symptoms related to the presence of an autoimmune disease in patients with ILD; also highlights the importance of testing for antibodies related to myositis in patients with ILD who do not present overt muscle or skin involvement, especially those with NSIP or OP pattern. Finally, we identify factors that would be related to functional improvement in those who receive immunosuppressive treatment in the context of MSA/MAA-ILD in an ethnically Latin American cohort.
Summary at a glanceIn this article we describe the clinical, radiological and pulmonary function of a cohort of Latin-American patients with ILD who are positive for myositis specific and associated antibodies; and the risk factors associated to pulmonary function at onset and long term follow up.
ContributionsAlberti María Laura: Conceptualization, Data curation, Investigation, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft (e) and Writing – review & editing (f).
Wolff Verónica: Conceptualization, Data curation, Investigation, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft (e) and Writing – review & editing (f).
Reyes Felipe: Conceptualization, Data curation, Formal analysis, Investigation, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft (e) and Writing – review & editing (f).
Juárez-León Ernesto: Conceptualization, Data curation and Writing – original draft (e).
Fassola Leandro: Data curation and Resources.
Carballo Gabriel: Resources and Writing – review & editing (f).
Buendía-Roldán Ivette: Data curation, Visualization and Writing – review & editing (f).
Rojas-Serrano Jorge: Visualization and Writing – review & editing (f).
Caro Fabián: Conceptualization, Data curation, Resources, Visualization and Writing – review & editing (f).
Florenzano Matías: Conceptualization, Data curation-Project administration, Visualization and Writing – review & editing (f).
Paulín Francisco: Conceptualization, Data curation, Formal analysis, Methodology, Visualization and Writing – review & editing (f).
FundingThis investigation has not received specific aid from public sector agencies, the commercial sector or non-profit entities.
Conflict of interestAll authors declare they have no conflicts of interest.


