






Although advances in biological medicine have seen significant progress in the treatment of autoimmune and inflammatory disease, many patients do not experience a satisfactory response. Hence, there are two challenges facing the medical research community. The first is to continue development in the field of existing biological therapies, such as monoclonal antibodies. The second is to open new frontiers of research and explore treatment alternatives for non-responders to other therapies. Attention has increasingly turned to the therapeutic potential of small molecule weight kinase inhibitors (SMKIs), currently used extensively in oncology and hematology. Initial research into the therapeutic value of SMKIs for autoimmune and inflammatory diseases has been encouraging. SMKIs are taken orally, which reduces cost for the health provider, and could increase compliance for the patient. This is why research is now focusing increasingly on SMKIs as a new generation line of treatment in these diseases. Tofacitinib, an inhibitor of Janus-kinase, is currently the only drug approved for the treatment of rheumatoid arthritis by FDA. However, much more needs to be done to understand the intracellular signaling pathways and how these might affect disease progression before solid conclusions can be drawn.
Pese a los avances terapéuticos en las enfermedades autoinmunes e inflamatorias, muchos pacientes no logran un control adecuado de la enfermedad. De ahí la necesidad de optimizar el uso de las terapias biológicas y de explorar nuevas opciones terapéuticas. La disponibilidad de fármacos que inhiben proteínas-cinasas ya es una realidad en especialidades como oncología y hematología, donde los resultados asociados a la evolución clínica de la enfermedad han sido prometedores. La principal ventaja de estos fármacos es la administración oral, que podría favorecer la adherencia del paciente y reducir los costes asociados al tratamiento. Tofacitinib, inhibidor de tirosinas-cinasas, actualmente es el único fármaco de esta categoría aprobado para el tratamiento de la artritis reumatoide por la FDA. Estas dianas terapéuticas son evaluadas actualmente en diversas enfermedades autoinmunes e inflamatorias. Sin embargo, el conocimiento y la comprensión de las vías de señalización intracelular siguen siendo limitados, persistiendo dudas en cuanto al mecanismo de acción, la eficacia y los posibles efectos secundarios asociados al uso de estos nuevos fármacos.
Until a little over 10years ago, the therapeutic options for the treatment of diseases such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), spondyloarthropathies (SA), psoriasis (PsO) and inflammatory bowel disease (IBD)—primarily Crohn's disease (CD) and ulcerative colitis (UC)—were very limited. At the present time, there is a wide spectrum of therapeutic options, including monoclonal antibodies to interleukins (IL), IL receptors, antigen recognition receptors and intercellular communication.1 Recently, protein kinases (PK) have emerged as therapeutic targets for the treatment of these diseases, which has led to the development of inhibitors that block the activity of these proteins.2 All this has contributed to deepen our knowledge of the immune mechanisms underlying the disease and the therapeutic response.
Protein Kinases: Definition and Function Relative to Autoimmunity and InflammationProtein kinases are enzymes that modify other proteins and/or enzymes biochemically, activating or deactivating them, depending on the objective of the intracellular communication from the membrane toward the nucleus. The sequencing of the human genome and advances in proteomics have enabled the identification of more than 500 PK, which have been cataloged in a system referred to as the “human kinome”.3 Depending on their cellular location and function, PK have been classified into 7 major groups, which include the tyrosine kinases (TK). Tyrosine kinases are enzymes that serve as mediators between the reception of an extracellular signal and the occurrence of an effector response. Their activation is produced by the phosphorylation or transfer of phosphate groups to the hydroxyl group of the tyrosine residues of the enzyme.4
The catalytic activity of PK is exhibited with: (a) redundancy: an action or response can be catalyzed by the simultaneous activation of separate enzymes; (b) pleiotropy: the activation of a single enzyme can promote different cell responses; and (c) synergy: the activation of several signaling pathways is necessary to achieve a specific effect and, thus, proper cell communication and function.
Yet, where within the framework of the immune system do PK fit in? The complex function of the cells of the immune system depends in part on the PK because of their decisive role in the cell signaling processes of growth, maturation, differentiation, migration, inflammation, aging and apoptosis. Thus, it is essential that PK activation be efficient, specific and appropriate, since defects in the activation and regulation of these mechanisms can affect the endocrine system and/or promote the development of malignant and/or anomalous cell phenotypes, as in cancer and autoimmunity.2,5,6
What Does the Intracellular Signaling Process Involve?As we mentioned above, the objective of intracellular signaling is to produce a nuclear response to the reception of a stimulus that is recognized by the cell membrane receptors. Protein kinases can form part of these receptors or be associated with the cell membrane, as they comprise 2 types: receptors with intrinsic kinase activity and receptors that have no intrinsic kinase activity but are associated with the kinases in their cytoplasmic domain.7
This signaling process can be summarized as follows:
- (a)
Recognition of different cytokines—chemokines, interleukins, growth factors or antigens—on the part of the specific receptors found on the cytoplasmic membrane.
- (b)
Binding of this receptor to the stimulus that induces conformational changes in the receptor and adjacent proteins.
- (c)
These conformational changes promote the activation of the kinases and, in the case of TK, the phosphorylation of its tyrosine residues.
- (d)
The activation of the kinases triggers that of other proteins to create a cascade directed toward the cell nucleus
- (e)
The start of gene transcription for protein synthesis, or the initiation of cellular processes such as cell differentiation, maturation, proliferation and apoptosis. This can be accompanied by the synthesis/activation of regulatory and/or inhibitory proteins, such as the phosphorylases which, in the case of TK, interrupts the intracellular signaling process by dephosphorylation (Fig. 1).
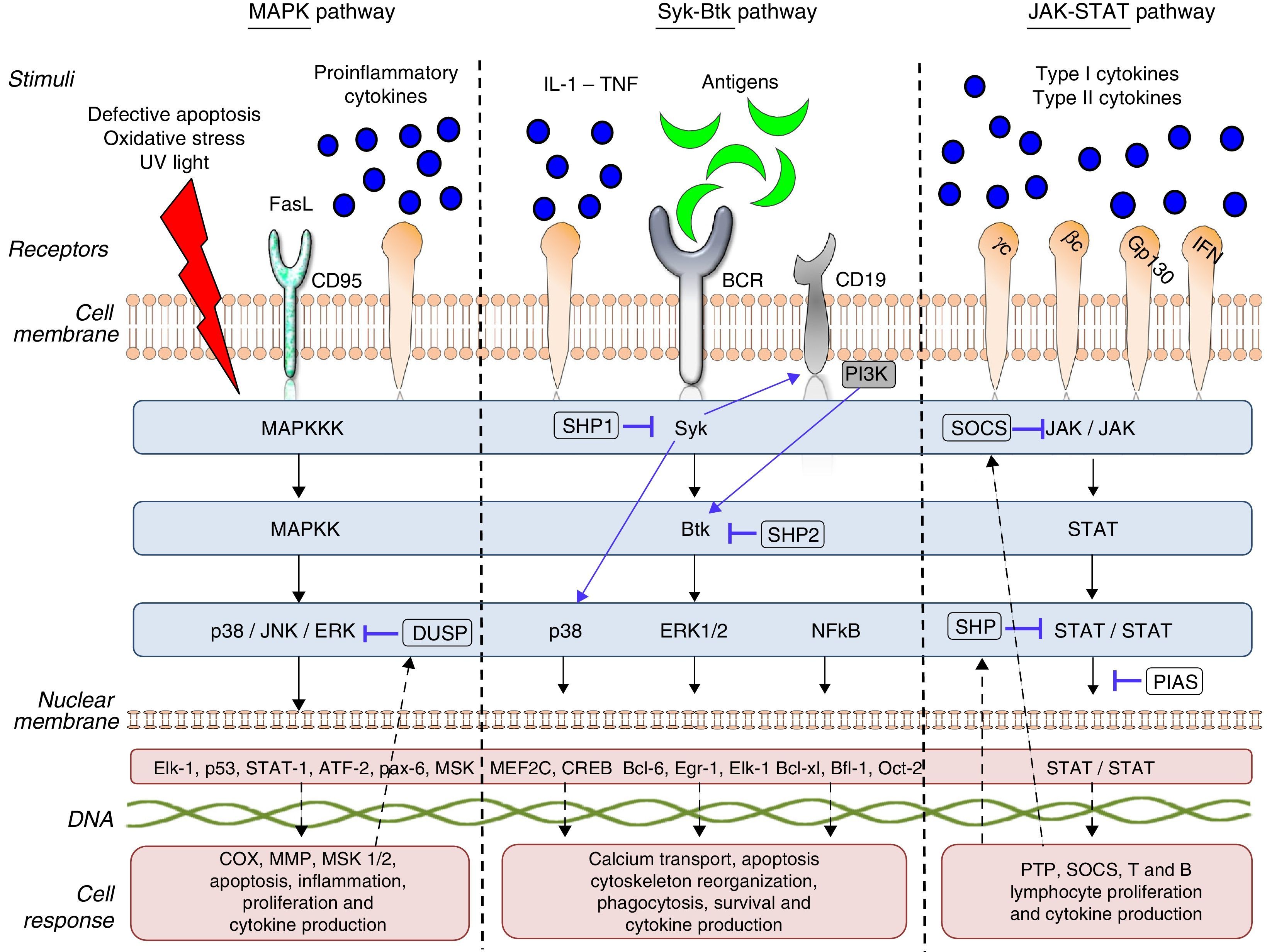
The cell communication system is made up of different “signaling pathways”, in which PK participate according to the objective of the signaling and the cell subpopulation they are in. To understand and describe the signaling process or cascade, the terms “upstream” and “downstream” are used. The literal translation of these 2 terms in Spanish would be “corriente arriba” and corriente abajo”, analogy of the flow of a current of water that is utilized to indicate the direction and position of the proteins within the intracellular signaling cascade. Thus, when we say that an action takes place upstream it means that it occurs in the direction of the cell membrane, like cell receptor recognition and binding, On the other hand, when we say that this action takes place downstream, it means that it occurs in the direction of the cell nucleus, like gene transcription. Thus, if we talk about a TK like Janus kinase 3 (JAK-3), any component that it activates and/or stimulates, such as the cytokines that bind to the common γ chain, is upstream (above it in the signaling cascade) and any component that the enzyme itself activates or regulates, as, for example, the activation and development of T lymphocytes and natural killer (NK) cells and their homeostasis is downstream (beneath it in the signaling cascade). Table 1 provides a detailed list of the meanings of these terms and the abbreviations or acronyms of each of the proteins, genes and transcription factors described in this review.
Abbreviations.
| AMA | American Medical Association | Sociedad Médica Americana |
| APhA | American Pharmacists Association | Sociedad Americana de Farmacéuticos |
| AS | Ankylosing spondylitis | espondilitis anquilosante |
| ATF-2 | Transcription factor 2 response to stress and DNA damage | factor de transcripción 2 que responde al estrés y al daño del ADN |
| ATP | Adenosine triphosphate | adenosina trifosfato |
| BCR | B cell receptor | receptor del linfocito B |
| Bcl-6 | B-cell lymphoma 6 protein | proteína 6 del linfoma del linfocito B |
| Bcl-xl | B-cell lymphoma-extra large | proteína del linfoma del linfocito B gigantes |
| Bfl-1 | BCL2 protein family from a human fetal liver | familia de proteínas BCL2 del hígado fetal humano |
| Bmx | Bone marrow tyrosine kinase gene in chromosome X protein | gen en el cromosoma X de la TC de la medula ósea |
| Btk | Bruton's tyrosine kinase or BPK (B cell progenitor kinase) | tirosina-cinasa de Bruton o cinasa del linfocito B progenitor |
| CD | Crohn's disease | enfermedad de Crohn |
| CD95 | Apoptosis antigen 1 or Fas receptor | antígeno apoptótico 1 o receptor de FasL |
| CHMP | Committee for medicinal products for human use | Comité de medicamentos para uso humano |
| COX | Cyclooxygenase | ciclooxigenasa |
| CREB | cAMP response element-binding protein | proteína de unión a la respuesta de cAMP |
| DNA | Deoxyribonucleic acid | ácido desoxirribonucleico |
| down-stream | Down-stream | señalización hacia abajo en la cascada |
| DUSP | Dual specificity protein phosphatase gene | genes que codifican proteínas-fosfatasas que inactivan p38, JNK y ERK |
| Egr-1 | Early growth response protein 1 | proteína de respuesta temprana al crecimiento 1 |
| Elk-1 | E-26-like protein 1 | proteína tipo E-26 |
| EMA | European Medicines Agency | Agencia Europea del Medicamento |
| Emt | Epithelial-to-mesenchymal transition | transición del epitelio mesénquimal |
| ERK | Extracellular signal-regulated kinase | PC que regula señales extracelulares |
| Etk | Endothelial and epithelial tyrosine kinase | TC endotelial y epitelial |
| FasL | Fas ligand or CD95 ligand | ligando del receptor de Fas o CD95 |
| FDA | Food and Drug Administration | Agencia reguladora de alimentos y medicamentos |
| GM-CSF | Granulocyte macrophage colony-stimulating factor | factor estimulante de colonias de granulocitos y monocitos |
| IBD | Inflammatory bowel disease | enfermedad intestinal inflamatoria |
| IL | Interleukin | interleucina |
| INN | International Nonproprietary Name | Programa internacional de nombres genéricos de la Organización Mundial de la Salud (OMS) |
| IFN | Interferon | interferón |
| ITAM | Immunoreceptor tyrosine-based activation motif | inmunoreceptores activadores que contienen tirosina |
| ITK | IL-2 Inducible T-cell Kinase | cinasa del linfocito T |
| ITP | Immune thrombocytopenic purpura | púrpura trombocitopénica autoinmune |
| JNK | c-Jun N-terminal kinase | PC que fosforila la proteína c-Jun |
| LPS | Lipopolysaccharide | lipopolisacáridos |
| Lyn | Lck/Yes related novel tyrosine kinase | TC asociadas Lck y Yes |
| MAPK | Mitogen activated protein kinase | PC activada por mitógenos |
| MAPKK | MAPK: mitogen activated protein kinase | PC activada por mitógenos que activan MAPK |
| MAPKKK | MAPKK: mitogen activated protein kinase | PC activada por mitógenos que activan MAPKK |
| MEF2C | Myocyte enhancer factor 2 | factor potenciador de miocitos 2 |
| MMP | Matrix metalloproteinase | metaloproteinasas |
| MSK-1/2 | Mitogen- and stress-activated kinase 1 and 2 | PC 1 y 2 activada por mitógenos y el estrés oxidativo |
| NFAT | Nuclear factor of activated T-cells | factor nuclear que regula la activación del linfocito T |
| NF-kB | Nuclear factor kappa-light-chain-enhancer of activated B cells | factor nuclear que regula la transcripción de las cadenas ligeras kappa (κ) |
| NK | Natural killer cells | linfocitos asesinos naturales |
| Oct-2 | Transcription factor 2 | factor de transcripción 2 |
| Pax-6 | Paired box protein 6 | proteína pareada 6 |
| PIAS | Protein inhibitor of activated STATs | proteína inhibidora de la transcripción de STAT |
| PIP 2 y 3 | Phosphatidylinositol bi and trisphosphate | fosfatidil inositol bi y trifosfato |
| PI3K | Phosphatidylinositol 3 kinase | PC de fosfatidil inositol 3 |
| PK | Protein kinase | proteína cinasa |
| PKC | Protein kinase C | proteína-cinasa C |
| PLC | Phospholipase C | fosfolipasa C |
| Pso | Psoriasis | psoriasis |
| PTEN | Phosphatase and tensin homolog | fosfatidilinositol-3,4,5-trisfosfato 3-fosfatasa |
| PTP | Protein tyrosine phosphatases | tirosinas-fosfatasas |
| RA | Rheumatoid arthritis | artritis reumatoide |
| RANKL | Receptor activator of NF-kB ligand | ligando del receptor activador del NF-kB |
| SHP-1 | Src homology region 2 domain-containing phosphatase-1 | fosfatasa 1 de las tirosinas-cinasas Src |
| RHOA | Ras homolog family member A | proteína GTPasa que regula la actina |
| SLE | Systemic lupus erythematosus | lupus eritematoso sistémico |
| SMKI | Small molecular weight kinase inhibitors | pequeñas moléculas inhibidoras de PC |
| SNP | Single nucleotide polymorphism | polimorfismo de solo un nucleótido |
| SOCS | Suppressors of cytokine signaling | supresor de la señalización de citocinas |
| Src | Proto-oncogene c-Src | PC que fosforila el protooncogen SRC |
| STAT | Signal transducer and activator of transcription | transductor de señal y activador de la transcripción |
| Syk | Spleen tyrosine kinase | tirosina-cinasa del bazo |
| TCR | T cell receptor | receptor del linfocito T |
| Tec | Tyr protein-kinases enzymes cytosolic | TC citosólica |
| TK | Tyrosine kinase | tirosina cinasa |
| TLR | Toll-like receptor | receptor tipo Toll |
| TNFα | Tumor necrosis factor alpha | factor de necrosis tumoral alfa |
| Tsk | T-cell signaling protein | proteína señalizadora del linfocito T |
| UC | Ulcerative colitis | colitis ulcerosa |
| up-stream | Up-stream | señalización arriba en la cascada de señalización |
| USAN | United States Adopted Names | Comisión americana encargada de asignar los nombres genéricos a nuevos fármacos |
| USP | United States Pharmacopeial Convention | Convención farmacopea de Estados Unidos de América |
| UV | Ultraviolet light | radiación ultravioleta |
| ZAP70 | Zeta-chain-associated protein kinase 70kDa | proteína asociada a la cadena zeta de 70 kilo-dalton |
There are 3 large subfamilies of mitogen-activated protein kinase (MAPK): p38, extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK). p38 is a serine/threonine kinase, and the best characterized subfamily to date: it has 4 homologous isoforms, alpha (α), beta (β), gamma (γ) and delta (δ), all of which are products of different genes that catalyze the same reaction. The α and β isoforms are ubiquitous, whereas the γ isoform is found in skeletal muscle and the δ isoform in the pancreas, small intestine and testes. On the other hand, ERK and JNK have 2 and 3 isoforms, respectively.8,9p38 and JNK are activated upstream by a number of stimuli, such as the Fas ligand (FasL), growth factors, proinflammatory cytokines, lipopolysaccharides (LPS), viral proteins and osmotic cellular stress in dendritic cells, neutrophils, macrophages and T and B lymphocytes. These enzymes regulate fundamental cellular processes that take place downstream, such as cell cycle regulation, apoptosis, cell aging and the production of cytokines such as IL-10. ERK is activated upstream by infectious agents, mitotic signals, growth factors, hormones and proinflammatory cytokines, and activates cytoskeletal protein synthesis, cell proliferation and cell differentiation downstream.8,10
This pathway is negatively regulated by MAPK kinases—MKK3 and MKK6—(MAPK-3 and 6) and by protein tyrosine phosphatases (PTP) of MAPK, like the so-called dual specificity protein phosphatase (DUSP). Likewise, IL1-RA and IL-10 production limits the production of proinflammatory cytokines and prostaglandins signaled by the pathway.11
Activated ERK, JNK and p38 are found in synovial tissue of patients with RA, demonstrating that they play an important role in inflammation and tissue damage in autoimmune diseases. Failures in the regulation of these signaling pathways induce changes in the innate and adaptive immune response, tumor cell proliferation, insulin resistance, and the development of neurological and/or degenerative diseases, failures in infection control and autoinmunidad.8,10 p38 is a key protein in the regulation of the proinflammatory response and, as such, was one of the first PK to be investigated as a therapeutic target in autoimmunity and inflammation.12
“Syk–Btk” PathwayThe Syk family comprises 2 members: zeta-chain-associated protein kinase 70 (ZAP70) and spleen tyrosine kinase (Syk).13 ZAP70 limits its expression to T lymphocytes and NK cells; in contrast, Syk is expressed on hematopoietic cells, mast cells and synoviocytes. Syk binds to the cytoplasmic region of receptors containing the immunoreceptor tyrosine-based activation motif [ITAM]), like macrophage Fcγ receptors, neutrophils, mast cells and T-cell receptor/B-cell receptor (TCR/BCR).14 In RA, it has been observed that Syk is activated in synoviocytes by proinflammatory cytokines like tumor necrosis factor (TNF) and IL-1, which induce JNK activation and IL-6 and metalloproteinase (MMP) expression upstream.15 Likewise, the activation of Syk promotes IL-12 and IL-13 synthesis, as well as the processes of cell proliferation, differentiation, survival, degranulation and phagocytosis.16
The Btk family has 4 members: Bruton tyrosine kinase/phosphoinositide 3-kinase (Btk/PI3K), IL-2 inducible T-cell kinase/epithelial-to-mesenchymal transition/tyrosine-protein kinase (Itk/Emt/Tsk), bone marrow tyrosine kinase gene in chromosome X protein/endothelial/epithelial tyrosine kinase (Bmx/Etk) and Tyr protein kinase cytosolic enzymes (Tec)17 Btk is a TK that is expressed in all the hematopoietic cells and lymphocytes except T lymphocytes and mature plasma cells. It is fundamental in lymphopoiesis and is downstream from Syk in this signaling pathway. Antigen presentation to BCR, IL-6 and erythropoietin activate upstream enzymes of the proto-oncogene c-Src family (Src), which phosphorylate Syk and, subsequently, Btk. Their phosphorylation initiates a number of cell processes, such as proliferation, survival, migration, angiogenesis, antigen presentation and cytokine production.18
PI3K is a subfamily of lipid kinases grouped from I to IV. They can be activated by the Syk–Btk axis by toll-like receptors (TLR) and adhesion molecules. Class I PI3K act on phosphatidylinositol bisphosphate (PIP2), generating a second messenger, phosphatidylinositol trisphosphate (PIP3), which binds to Btk, inducing its phosphorylation by Syk and Lyn. These enzymes play an important role in leukocyte migration, mast cell degranulation and entry of calcium into the cell.19
Downstream activation of these enzymes promotes the activation of other pathways that regulate inflammation, like MAPK (p38, ERK1/2), phosphoinositide 3-kinase (PI3K), phospholipase C (PLC) and transcription factors, such as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) and nuclear factor of activated T-cells (NFAT). NF-kB is an important protein complex comprised by transcription factors that are implicated in a large number of cell processes that are indispensable for cell survival and function, and NFAT regulates B lymphocyte activation, differentiation and maturation.20
Tyrosine phosphatases, like Src homology region 2 domain-containing phosphatase (SHP)-1, SHP-2, protein kinase C (PKC) and phosphatase and tensin homolog (PTEN), regulate the enzyme activity of Syk, Btk and PI3K in cellular processes such as apoptosis, adhesion, migration and proliferation. Mutations in the Btk gene, like X-linked agammaglobulinemia (XLA), can block B lymphocyte differentiation and maturation, leaving the cells dysfunctional for antigen presentation. Likewise, the constitutive activation of this enzyme and signaling failures in the BCR–Btk axis promote the development and survival of aberrant B lymphocyte phenotypes. In murine lupus models, PI3K-deficiency has been found to reduce CD4+ T lymphocyte survival, antibody production, TNF-α, proteinuria and glomerulonephritis.21 Failures in the regulation of these enzymes can induce antibody-mediated and allergic diseases, like asthma and allergic rhinitis, as well as autoimmunity due to loss of tolerance and defects in antigen presentation by B lymphocytes, facilitating the development of diseases like RA, leukemia and cancer.22–24
“JAK–STAT” PathwayTo date, 4 members of the JAK family have been identified: JAK-1, JAK-2, JAK-3 and TYK-2, which are ubiquitously expressed in all cells, with the exception of JAK-3, which is confined to hematopoietic cells. These TK are stimulated upstream by type I and type II cytokine receptors. JAK-1 is activated by cytokines that bind to receptors containing the common γ chain and transmembrane glycoprotein 130; JAK-2, by receptors containing transmembrane glycoprotein 130, IL-3 and interferon (INF)-γ; JAK-3, by receptors containing the common γ chain; and, lastly, TYK-2 is stimulated by IL-12 and LPS.25 These enzymes are structurally associated with the cytoplasmic region of cytokine receptors in the form of dimers or trimers. Each combination of JAK and/or TYK is modulated by specific stimuli and has a different function in cell signaling and, consequently, in the immune system. The dimers JAK-1/JAK-3, JAK-1/TYK-2, JAK-1/JAK-2 and JAK-2/TYK have different downstream functions characteristic of innate and adaptive immunity, whereas others, like the dimer JAK-2/JAK-2, regulate the maturation and differentiation of hematopoietic cell lines.26,27
JAK/TYK phosphorylation induces the phosphorylation of the transcription factors, signal transducers and activators of transcription (STAT). These factors constitute a family with 7 members: STAT 1, 2, 3, 4, 5a, 5b and 6, which are found downstream in the signaling cascade and are associated with homodimers or heterodimers that, having been phosphorylated by JAK, dimerize and translocate to the nucleus, where they bind to the target genes, increasing or repressing gene transcription and cell function.28
Mutations in these enzymes or defects in the signaling have been associated with the development of myeloproliferative, autoimmune and inflammatory disorders.29,30 Genomic association studies have described the role of certain receptor, cytokine and enzyme genes associated with each of these JAK/TYK dimers and/or trimers and the susceptibility to developing allergies and different autoimmune and inflammatory diseases. For example, the signaling of the dimer JAK-1/TYK-2 by IFN-α is associated with Pso and IBD; JAK-2/TYK-2 with IL-12 and IL-23 in Behçet's disease, RA, SLE, Pso, IBD and AS; and the trimer JAK-1/JAK-2/TYK-2 with IL-6 stimulation and susceptibility to developing IBD.28
This signaling pathway is regulated by different proteins, such as suppressors of cytokine signaling (SOCS), protein inhibitor of activated STAT (PIAS) and/or the tyrosine phosphatases SHP-1 and SHP-2, which block the binding of the STAT to DNA. This signaling pathway also regulates and is regulated by other signaling pathways, such as PI3K, MAPK and NF-kB.25,31
Challenges of the Development of Tyrosine Kinase InhibitorsAutoimmune and inflammatory diseases are a group of chronic diseases in which, despite the availability of a number of therapeutic options, a percentage of patients do not respond adequately, probably due to the complexity of the immune system, as well as to the characteristics of each disease, of the drug and of the patient him- or herself. For all these reasons, it is still a challenge to select the appropriate therapeutic targets for the development of new drugs that increase the therapeutic options when the conventional and biologic therapies presently approved have failed. In recent decades, we have seen the development of a number of monoclonal antibodies to cytokines and cytokine receptors characteristic of the extracellular matrix or located in the cell membrane. For this reason, and due to the fact that JAK–STAT are pleiotropic enzymes that participate in the intracellular signaling of multiple cytokines, TK inhibitors have been developed as a new, more specific and effective therapeutic option in the inhibition of each disease process.26
The main advantage of these small molecular weight kinase inhibitors (SMKI) is their oral administration versus the subcutaneous or intravenous administration of the monoclonal antibodies. This administration route facilitates adherence and increases the willingness of patients to receive the treatment, and could reduce costs with respect to the existing drugs. This group of drugs is absorbed in the stomach and small intestine. In the liver, the cytochrome P450 system takes charge of its metabolism, in which the presence of genetic polymorphisms like the single nucleotide polymorphism (SNP), can alter the pharmacokinetics, bioavailability and efficacy of the active ingredient, as has been reported with another type of drugs metabolized by this system. The TK inhibitors block the site for the binding of TK to adenosine triphosphate (ATP). This point is critical in the development of these new drugs because it is indispensable that the block be selective; for the inhibition to be effective and specific, and to prevent the inhibition of multiple TK and an increase in the risk of toxicity and/or of secondary effects.
This group of drugs has been included in the category of antineoplastic agents and has been assigned the suffix -itinib (International Nonproprietary Name [INN]).32 The nomenclature utilized for assigning generic names follows the recommendations of an advisory committee of the American Medical Association and the American Pharmacist's Association (USAN, AM, USP, APhA).33
Use and Perspectives of Tyrosine KinaseThe clinical success of selective tyrosine kinase inhibitors in the treatment of tumors and leukemias (colorectal, gastric and breast cancer, hepatocellular carcinoma, sarcoma, metastatic melanoma, acute lymphoblastic leukemia and chronic myeloid leukemia) approved by the United States Food and Drug Administration (FDA) and the European Medicines Agency (EMA) has led to an increase in preclinical and clinical trials and studies of therapeutic options for the treatment of autoimmune and inflammatory diseases (www.clinicaltrials.gov). It has also deepened our understanding of the role of TK in intracellular signaling and inflammation.34
In murine models of RA treated with p38 (SB203580 and PH-797804), JNK (SP600125) and ERK (PD98059), a decrease in proinflammatory factors, such as prostaglandins, and in IL-10 and joint inflammation has been observed.35,36 A recent study reports that SP600125 inhibits MMP expression, reduces cartilage loss and increases the number of regulatory T lymphocytes, also in a murine model of RA.37 Due to the limited efficacy observed with the use of JNK and ERK inhibitors, the existing studies focus on its use in autoimmune diseases. In IBD, the utilization of p38 inhibitors such as semapimod (Ferring Pharmaceuticals) reduces inflammatory activity in CD, although the response is less than satisfactory in severe forms.38 Tests with the drug doramapimod (Boehringer Ingelheim) in RA, AS and CD resulted in a decrease in the production of proinflammatory cytokines by certain cell lines.39,40 Treatment with pamapimod (Hoffman-La Roche) was found to have little efficacy in RA, and was accompanied by a high rate of infections and skin conditions.41,42
With respect to the inhibition of Syk, studies in vitro and in animal models of RA showed that, PRT062070 (cerdulatinib), an inhibitor of Syk and JAK-1/3, reduced inflammatory activity and synovitis resulting from the suppression of autoantibody production caused by the blocking of signaling and B lymphocyte activation.43 A study involving 2 models of RA in mice treated with P505-15, a selective Syk inhibitor, corroborated its efficacy in blocking BCA signaling and activation and in inhibiting basophil activation via Fcγ.44 In clinical trials, fostamatinib disodium (AstraZeneca-Rigel), a Syk inhibitor, is being evaluated in patients with SLE, Pso and autoimmune thrombocytopenia purpura. In animal models of RA, the prodrug fostamatinib (R-788) was shown to reduce proinflammatory IL and MMP production and delay joint destruction, probably due to its acting on T lymphocytes and osteoclasts.45 On the other hand, it was seen to prevent renal damage and the development of skin lesions in a murine model of lupus.46 However, despite these findings, and in a preliminary phase III study, fostamatinib was not superior to placebo in patients with RA who had had a poor response to biologic therapies.47
Different Btk inhibitors, like dasatinib (Bristol-Myers Squibb), PCI-32765 (ibrutinib, Janssen Research & Development, LLC), CC-292 (azacitidine, Celgene Corporation) and GDC-0834, have been approved for the treatment of multiple myeloma, lymphoma and B-cell leukemia and are being evaluated in preclinical and clinical studies of RA and SLE. In vitro studies have demonstrated that inhibition of Btk reduces bone loss in murine models of receptor activator of NF-kB ligand (RANKL)-induced osteoporosis, as well as autoantibody production, and delays the development of kidney disease in a murine model of lupus.48–50 In a model of collagen-induced arthritis in rats, GDC-0834 promoted a reduction of joint inflammation.24,51 Moreover, in a murine model of lupus, a decrease in IgG deposits and in cell infiltrate and activation was observed when the mice were treated with the inhibitor RN-486.52
PI3K inhibitors are being widely studied in hematopoietic disorders and solid tumors. In preclinical studies, molecules such as IC-87114 and CZC24832 reduced IL-17 production, blocking Th17 lymphocyte differentiation and osteocyte generation in murine models of RA. Another inhibitor of PI3K, AS-605240, reduced inflammation, neutrophil infiltrate and erosions. This same molecule increased survival and reduced autoantibody production and attenuated glomerulonephritis in a murine model of lupus.19
The results obtained with different molecules directed against the JAK enzymes have shown promise in different clinical trials for the treatment of Pso and RA, and the second generation of these molecules is defined as being more specific and effective as inhibitors. In models of RA treated with decernotinib (Vertex Pharmaceuticals Inc.), a molecule directed against JAK-3, there was a reduction in joint inflammation,53 although preliminary clinical results revealed an increase in the risk of infections and in the transaminase levels.54 ASP015K (Astellas Pharma Inc.), a JAK-3 inhibitor that, to a lesser extent, inhibits JAK-1 and JAK-2 as well, has promoted the reduction of the epidermis thickness and of cell proliferation in patients with Pso.55 Baricitinib (Incyte and Eli Lilly Company), a JAK-1/JAK-2 inhibitor, has been found to reduce inflammatory activity and favor the preservation of cartilage and bone in a murine model.56 Ruxolitinib (Incyte Corporation and Novartis) is an oral drug that inhibits JAK-1/JAK-2; approved for the treatment of proliferative disorders, its topical form is being evaluated for use in the treatment of Pso. The study of this molecule in murine models showed that it promoted a decrease in STAT-3 phosphorylation and, with that, the reduction of edema, lymphocyte infiltration and psoriatic plaques.57 Results in clinical trials indicated that this drug is well tolerated and is effective in the treatment of Pso.58 On the other hand, tofacitinib (Pfizer) is the only TK inhibitor that is commercially available in the United States and other countries for the treatment of RA. It is a functional inhibitor specific for JAK-3 that also inhibits JAK-1 and, to a lesser extent, JAK-2.59,60 In preclinical studies, this inhibitor was found to reduce the levels of chemokines, interleukins, acute phase reactants and RANKL, promoting the reduction of bone reabsorption mediated by osteoclasts and inflammation.61 It has also been seen to inhibit the proinflammatory response of Th1 and Th1762 and prevent cartilage damage in animal models.63
In more recent studies, it has been observed that, in RA patients treated with tofacitinib, there is an inhibition of the proliferation of CD4+ T lymphocytes without affecting their absolute number on the periphery. Moreover, it has been found that, before treatment, a small number of CD8+ T lymphocytes are correlated with the development of secondary effects during the treatment in patients with RA.64 Given the efficacy of tofacitinib in the treatment of RA, phase III clinical trials are now underway in Pso, as well as UC, CD and AS.
A summary of the major clinical trials carried out in autoimmune and inflammatory diseases is provided in Table 2. Despite the challenge posed by the development of these inhibitors when compared with monoclonal antibodies—that is, intracellular proteins versus extracellular proteins, the secondary effects reported do not seem to differ from those observed up to now with biologic therapies: neutropenia, high rates of infection, hepatotoxicity and elevated liver enzymes, altered thyroid function, fatigue, hypertension, skin rashes, delayed wound closure, myelosuppression, diarrhea, elevated creatinine levels and hyperlipidemia, among others. For this reason, work is currently underway to develop inhibitory molecules with greater specificity and an effect better aimed at each disease.
Protein Kinase Inhibitors Evaluated for the Treatment of Autoimmune and Inflammatory Diseases.
| Therapeutic target | Drug/molecule | Laboratory | Clinical trial | Indication | Status |
|---|---|---|---|---|---|
| p38 | Dilmapimod/SB-681323 | Glaxo Smith Kline | II | RA | Terminated |
| Semapimod/CNI-1493 | Ferring Pharmaceuticals | II | CD38 | Terminated | |
| Pamapimod/RO4402257 | Hoffman-La Roche | II | RA42 | Terminated | |
| PH-797804 | Pfizer | II | RA35 | Terminated | |
| VX-702 | Vertex Pharmaceuticals Inc. | II | RA65 | Terminated | |
| BMS-58294966 | Bristol-Myers Squibb | I | Pso | Terminated | |
| II | RA | ||||
| Talmapimod/SCIO-469 | Scios Inc. | II | RA67 | Terminated | |
| ARRY-371797 | Array BioPharma | II | RA and AS | Terminated | |
| Doramapimod/BIRB-796 | Boehringer Ingelheim | IIa | RA, Pso and CD39 | Terminated | |
| Syk | Fostamatinib disodium/R935788 | AstraZeneca-Rigel | II | SLE | |
| III | RA47,68 | Suspended | |||
| III | ITP | Recruiting | |||
| Btk | Azacitidine/CC-292 | Celgene Corporation | II | RA | Recruiting |
| JAK-1 | GSK2586184 | Glaxo Smith Kline | I | UC | Suspended |
| II | SLE and Pso | Terminated | |||
| Filgotinib/GLPG0634 | Galapagos NV. | II | RA, CD69 | Recruiting | |
| PF-04965842 | Pfizer | II | Pso | Recruiting | |
| JAK-2 | AC430 | Ambit Biosciences Co. | I | RA | Terminated |
| JAK-3 | ASP015K/JNJ-54781532 | Astellas Pharma Inc. | II | RA and Pso54 | Recruiting |
| Decernotinib/VX-509 | Vertex Pharmaceuticals Inc. | II and III | RA53 | Terminated | |
| JAK-3 and Syk | R-333 | Rigel Pharmaceuticals | II | SLE | Terminated |
| JAK-1/2 | Ruxolitinib/INCB-018424 | Incyte Co. and Novartis | II | RA and Pso58 | Terminated |
| Baricitinib/INCB-28050-LY-3009104 | Incyte and Eli Lilly Co. | II | Pso | Terminated | |
| III | RA70 | Recruiting | |||
| JAK-1/3 | Tofacitinib/CP-690-550 | Pfizer | Approved by FDA | RA59,60 | Recruiting |
| II | AS and CD71 | Recruiting | |||
| III | Pso and UC72 | Recruiting | |||
| JAK-2/3 | Lestaurtinib/CEP-701 | Cephalon | II | Pso | Terminated |
In November 2012, the FDA granted approval to tofacitinib (Xeljanz®) for the treatment of moderate to severe RA in adults who have had an inadequate response or intolerance to methotrexate (MTX). According to its technical specifications, tofacitinib is administered orally, at doses of 5mg twice daily, as monotherapy or in combination with MTX or other synthetic drugs. It should not be used in combination with biologic therapies or immunosuppressive agents such as azathioprine and cyclosporine. At the present time, in addition to the United States, tofacitinib has been approved for the treatment of RA in several countries, including Japan, Russia, Canada and Switzerland.
As for the European Community, in April 2013, the Committee for Medicinal Products for Human Use (CHMP) of the EMA considered that, despite the evidence of a reduction in the signs and symptoms of RA and an evident improvement in the physical function of these patients, the benefit was not sufficient to outweigh the associated secondary effects, such as serious infections, gastrointestinal perforations and cancer, despite the fact that the dossier presented to the FDA was the same as that presented to the EMA. Therefore, due to the uncertainty caused by the magnitude of the risks and their management in clinical practice, it was considered that making it commercially available was not worthwhile in terms of the benefits obtained with the treatment (EMA/CHMP/425279/2013, Procedure No. EMA/H/C/002542/0000). In view of this, the company requested a revaluation of the risks/benefits of the drug, and the group of experts, in accordance with article 12 of Regulation (EC) no. 726/2004, ruled that the risk/benefit ratio of tofacitinib was inadequate or had not been convincingly demonstrated and, thus, refused once again to authorize that it be marketed (EMA/CHMP/425279/2013). However, since this drug has been commercially available in the United States for 2 years, we hope that we will soon have evidence from clinical practice to corroborate the results obtained in the clinical trials. On the other hand, tofacitinib continues to be evaluated in different clinical trials for its use in other conditions involving autoimmunity and inflammation, to take position as a new therapeutic option in the treatment of this group of diseases.
ConclusionThe use of drugs that inhibit PK, like TK, could change the therapeutic approach in diseases involving failures in the regulation of the immune system, due to the ubiquity of these molecules in the process of intracellular signaling and, thus, in their effect on autoimmune and inflammatory processes. This could benefit those patients in whom multiple treatments have failed because of a loss of efficacy and secondary effects. Likewise, the introduction of these molecules that are administered orally would change patient adherence and reduce the cost associated with the treatment compared to the administration of monoclonal antibodies. However, the anything but promising results with some of these PK inhibitors have shown us that not all the intracellular signaling pathways are susceptible to being blocked to provide beneficial clinical effects. As new results in clinical practice concerning the use of these therapeutic targets are obtained, it will be possible to identify those signaling pathways that are most appropriate for the goal of obtaining a proper balance between the desired clinical efficacy and the adverse effects. In this respect, and in light of the evidence that is being obtained with the use of inhibitors of the JAK kinases, like tofacitinib, the JAK–STAT pathway is seen as one of the main therapeutic targets for the development of new drugs and the treatment of diseases of the immune system.
Ethical DisclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that no patient data appear in this article.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflicts of InterestLara Valor has received professional fees from Abbvie, Bristol-Myers Squibb, Pfizer, Roche Farma, UCB Pharma and Merck Sharp & Dohme Limited (MSD).Diana Hernández-Flórez declares that she has no conflicts of interest.
The authors thank Drs. Luis Carreño Pérez, Francisco Javier López Longo (Department of Rheumatology, Hospital General Universitario Gregorio Marañón, Madrid, Spain) and Gustavo Centeno Soto (Department of Clinical Pharmacology, Hospital Puerta de Hierro, Madrid, Spain) for their critical reading of this manuscript.
Please cite this article as: Hernández-Flórez D, Valor L. Los inhibidores de las proteínas-cinasas en enfermedades autoinmunes e inflamatorias: presente y futuro de nuevas dianas terapéuticas. Reumatol Clin. 2016;12:91–99.


