




Pyle's disease (OMIN number 265900) is a metaphyseal dysplasia of benign course, inherited with an autosomal recessive pattern. Some 30 genuine cases have been described so far. The cause of this process has been known since 2016, when its relationship to mutations in the gene encoding the sFRP protein, a known inhibitor of the Wnt pathway, was discovered. We report the case of a 58-year-old man, diagnosed with Pyle's disease based on his clinical and radiographic characteristics, whose phenotype suggested a differential control of cortical and trabecular bone homeostasis.
La enfermedad de Pyle (OMIN número 265900) es una displasia metafisaria de curso benigno que se hereda con un patrón autosómico recesivo. Se han descrito unos 30 casos genuinos hasta el momento. La causa de este proceso se conoce desde 2016, cuando se descubre su relación con mutaciones en el gen que codifica la proteína sFRP, un conocido inhibidor de la vía Wnt. Se presenta el caso de un varón de 58 años, diagnosticado de enfermedad de Pyle con base en sus características clínicas y radiográficas, cuyo fenotipo muestra un control diferencial de la homeostasis del hueso cortical y trabecular.
Pyle's disease (PD, OMIN number 265900) is a metaphyseal dysplasia of benign course,1 inherited with an autosomal recessive pattern.2 It was described in 1931 by Edwin Pyle,3 an orthopaedic surgeon from Connecticut (U.S.A.), on a 5 year old boy who presented with severe genu valgum in both knees and cranial deformities. Since then some 30 genuine cases have been described in different countries and continents. In a review made in the data bases of PubMed and Embase we only found one published case in Spain.4
Despite its rarity clinical interest in PD has recently risen on discovering its relationship with mutations in a soluble inhibitor of the Wnt5–7 pathway. Analysis of the phenotype of these patients and the events observed in mouse models which reproduce the dysfunction of the gene sFRP reveal a differential control in the homeostasis of cortical and trabecular bone. We now present a case with distinctive images of the cortical and trabecular bone and the relevant interest of the clinical symptoms of these patients is commented upon.
Clinical observationWe present the case of a 58 year old man who was sent to the outpatient rheumatology department on suspicion of bone fragility. At 6 years of age he had undergone surgery for marked bilateral genu valgum and at the time there was a suspicion of congenital osteopathy. The operation improved the statics of his lower limbs and the patient was able to have a normal life, with no relevant pathologies until 57 years of age, when, after a low intensity trauma, he suffered a fracture to the left collarbone and acetabulum. In the medical file no risk factors of osteoporosis or family history or personal history of interest were recorded, except for the before-mentioned surgical intervention. Physical examination revealed an adult with a good general appearance, with no craniofacial deformities, a moderate bilateral genu valgum and the limitations of the recent fractures, for which surgery was required. He was 182cm tall, weighed 81kg (BMI 24.45kg/m2), with nothing remarkable in the rest of the physical examination. General analyses tested normal, with ionic serum calcium of 2.41mmol/l, total alkaline phosphatase of 115u/l, and 25-OH-D3 of 32.8ng/ml and PTHi of 56pg/ml. Bone densiometry showed a T-score of −1.4 SD in lumbar spine (928mg/cm2) and −2.2 SD in neck of the femur (625mg/cm2). A radiographic study revealed a pattern which is characteristic of PD, with a metaphyseal enlargement of the tubular bones (Fig. 1), marked cortical thinning and normal epiphyseal morphology.
Discussion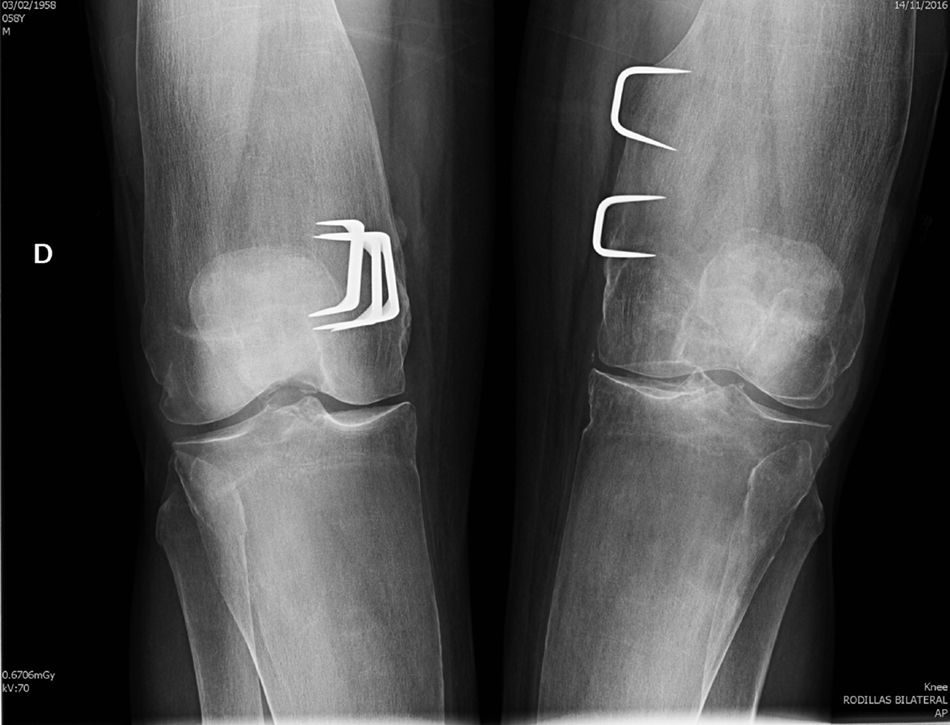
Our patient met with the radiographic characteristics of PD in the adult, which include a metaphyseal expansion of the long bones and a marked cortical thinning.8,9 All the long bones may be affected, although the characteristic findings are most obvious in the proximal tibia and in the distal femur (with the typical appearance of the “Erlenmeyer flask”) The craniofacial bones may be affected by discreet and occasional changes, such as patched osteosclerosis and obliteration of paranasal passages. However, when these alternations are marked they should rule out other hereditary disease, especially craniometaphyseal dysplasia, which shares with PD abnormal metaphyseal modelling, but which is clearly differentiated because the affected patients present a very unusual facial features, with hypertelorism and thickening of the nasal bridge, and there is no involvement of collarbone, ribs and ischium pubis branches. Camurati–Engelmann disease is also included in the group of metaphyseal dysplasia's but, unlike Pyle's disease, it causes hyperostosis at the skull base and cortical thickening in the long bones. Other processes to consider in the differential diagnosis of PD are osteopetrosis, where apart from the increase in bone density which characterises the group, images of “bone within bone” are appreciated, both in the vertebral bodies and in the diaphyseal and metaphyseal regions of the tubular bones. Other diseases to take into account in the differential diagnosis are Guacher's disease, Niemann–Pick's disease and the Thalassemias disorders, although these are easily ruled out by the medical file and additional tests, and also by the absence of extraosseous compromise and normal lab test results observed in Pyle's disease.
Fragility fractures are frequent in PD and are located in the metaphysis with thinned cortical bones and with a special preference in transition locations between the metaphyseal dysplasia bone and the diaphyseal bone, as may be observed in the collarbone fracture of our patient. In PD cortical architecture is affected, but the trabecular bone is normal and its density may even be increased. This surprising phenotype profile induces one to speculate with differentiated homeostasis between the cortical compartment and the trabecular compartment, the molecular origin of which has remained hidden until now and which we will briefly review below.
In 2016, Simsek Kiper et al.5 carried out a complete exome sequencing on 4 patients with Pyle's disease, identifying a bialelic “nonsense” mutation in gene sFRP4 (606570.0001) which encodes protein relating to Frizzled segregated 4 (sFRP4), a known inhibitor of the Wnt pathway. This defect was confirmed by another 2 groups almost simultaneously.6,7 In one confirmatory mouse model5 the sFRP4 gene was cancelled, and a progene with very thinned corticals was obtained in both the long and flat bones from a very early age, and an increase in trabecular volume and in the dynamic parameters of trabecular formation, with a reduction in the rate of endocortical and subperiostic apposition. The number of osteoclasts was normal in the trabecular bone, but had risen in the diaphyseal endocortical surface area. All findings are characteristic of human Pyle's disease and show that the dysfunction of the sFRP4 lead to an increased trabecular bone (as occurs in other diseases with release of the Wnt pathway10), but without warning, a reduced cortical thickness and fault in the cortical modelling during growth, which led to a metaphyseal widening and bone fragility which remained in the adult.
The Wnt pathway is the main signalling system to control tissue renewal.11,12 Its members act on a local level using specific tissues, directing the fate of progenitor cells. It is strongly regulated by a wide number of effectors that act as agonists or antagonists on an intracellular level, modulating the transduction of the signal, or on an extracellular level, regulating the ligand-receptor interactions.13 The sFRP family of proteins is the most numerous among the inhibitors segregated from the extracellular Wnt signal. In humans there are 5 known members (sFRP1-5). All of them share a similar structure domain to the cystein-rich binding domain to Wnt of the frizzled receptors, which facilitate the sequestration of the Wnt ligands, preventing them from binding to the receptor. There are tests of an alternative inhibition model through which the sFRP would form non functional complexes with these receptors, preventing their binding to the Wnt ligand.14 Through both systems, the sFRP may inhibit both the canonical and non canonical pathway of activating the Wnt signal, participating very definitively in osseous modelling and remodelling in the postnatal stage, the dysfunction of which reaches its maximum clinical expression in Pyle's disease.15 In the process, the trabecular bone presents with a similar phenotype to others which are characterized by an increase in the canonical Wnt signal. However, the cortical bone shows marked thinning, already present from early stages of skeletal maturity – a finding which is contrary to that expected when we liberate the Wnt signal of one of the main inhibitors and which show that the trabecular bone and the cortical bone have differential homeostatic regulation and control mechanisms.
Conflict of interestsThe authors have no conflict of interests to declare.
Please cite this article as: Arboleya L, Queiro R, Alperi M, Lorenzo JA, Ballina J. Enfermedad de Pyle: un modelo humano de homeostasis corticotrabecular diferenciada. Reumatol Clin. 2020;16:56–58.


