





Scleroderma is a rare disease with limited data in Latin America. Preliminary genetic studies suggest a strong African ascendance in the Dominican Republic, which could modulate the expression of the disease. The objective of this study is to describe the clinical and demographic characteristics of scleroderma in a series of 26 Dominican patients.
Materials and methodsPatients who fulfilled the EULAR/ACR criteria for scleroderma were selected from the Rheumatology Department of a tertiary health center; systemic sclerosis subtypes were defined according to the EULAR classification. Clinical and demographic information was obtained retrospectively from clinical records.
ResultsMean age at time of onset was 32.6±15 years; 68% of patients had 40 years of age or less. 73% of patients were females, with a female:male ratio of 2.7:1. The most affected systems were pulmonary and gastrointestinal; renal affection was scarce. Anti-Scl-70 antibodies were positive in 64% of patients, sometimes in coexistence with anti-centromere antibodies.
ConclusionsThe prevalence of systemic sclerosis is lower in the Dominican population than was reported elsewhere. The age of onset of the disease seems to be lower in the Dominican population than that reported in literature. A different pattern of autoantibodies is observed in this population.
La esclerodermia es una enfermedad rara, de la cual existe información limitada en América Latina. Estudios preliminares en genética muestran que la ascendencia de República Dominicana tiene fuerte influencia africana, lo cual podría modular la expresión de la enfermedad. El propósito de este estudio es describir las características clínicas y demográficas de esclerodermia en una serie de pacientes dominicanos.
Materiales y métodosSe seleccionaron los pacientes que cumplieron con los criterios del EULAR/ACR para esclerosis sistémica de la base de datos del servicio de reumatología de un centro terciario. Se definieron los subtipos de esclerodermia de acuerdo a la clasificación EULAR. La información clínica y demográfica al momento del diagnóstico fue obtenida de forma retrospectiva de los expedientes médicos.
ResultadosLa prevalencia fue estimada de 9,3 por millón de habitantes. Veinte y seis pacientes entraron al estudio. La edad media al momento del primer síntoma fue 32,6±15 años; el 68% de los pacientes tenía 40 años de edad o menos cuando aparecieron los síntomas. El 73,1% de los pacientes fue de sexo femenino, con una relación mujer:hombre 2,7:1. Los sistemas orgánicos más afectados fueron el pulmonar y el gastrointestinal; la afectación renal fue rara. Los anticuerpos anti-Scl-70 se encontraron positivos en el 64% de los casos y, en 2 casos, en coexistencia con anticentrómero.
ConclusionesLa prevalencia de esclerosis sistémica es menor en la población dominicana que la reportada internacionalmente. La edad de inicio de la enfermedad parece ser menor en la población dominicana que la reportada en la literatura. Un patrón distinto de autoanticuerpos es observado en esta población.
Scleroderma is a disease characterized by dysfunction of the peripheral vasculature, resulting in narrowing and eventual obliteration of the microvasculature, due to an aberrant immune system activation and fibrosis dependent of growth factors.1,2 Although the basic biological mechanisms of development and progression of scleroderma have still not been fully described,3 the reality is that several demographic factors appear to affect the incidence, prevalence and severity of this disease.4 In particular,5 it has been documented that black patients with scleroderma have a more aggressive and earlier clinical and serological profile than whites.
African ancestry is prevalent in much of Latin America,6 reaching 85% of influence on the genetics of the population of the Dominican Republic.7 However, in the Latin American population, there is limited information on scleroderma and, to the best knowledge of the authors, is absent in the Caribbean Antilles. The objective of this study is to describe the demographic and clinical characteristics of the disease in a number of Dominicans patients.
Materials and methodsPatientsPatients were seen at the Rheumatology department of the Hospital Regional Universitario José María Cabral y Baez in the period between March 2002 and May 2013 (n=1.609); we selected those that met the EULAR/ACR 2012 classification criteria for systemic sclerosis (SS),8 and who were compiled in the Dominican population EUSTAR database. For classification into subtypes we followed the recommendations of EULAR/ACR criteria for classification of SS and localized disease subtypes.9,10Fig. 1 summarizes the classification.
. Subcommittee for scleroderma criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee 9 to Kreuter et al.10")
Overall scleroderma classification. Adapted from: Preliminary criteria for the classification of systemic sclerosis (scleroderma). Subcommittee for scleroderma criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee 9 to Kreuter et al.10
Patients with overlap (n=1) syndromes, including mixed connective tissue disease (n=3) were excluded from this study. SS patients were divided into those with the diffuse form of the disease and those with the limited form (which included CREST syndrome); there were no patients with SS sine scleroderma. Patients of all age ranges were included because there is no pediatric rheumatology service in the region. Clinical and demographic information was obtained retrospectively from the department database and medical records; information at diagnosis was used. High resolution computed tomography (HRCT) and pulmonary function tests for the detection of interstitial lung disease were performed.
All patients signed informed consent; in the case of minors, the legal representative signed the informed consent, with the minor also signing the informed consent. The protocol was approved by the institutional ethics committee.
VariablesThe demographic variables analyzed were gender and age, both at diagnosis and at the time of onset of related symptoms. The age at diagnosis was calculated using the date on which the final diagnosis was made and the date of birth of the individual; the age at onset of symptoms was obtained through directed interrogation.
Regarding the disease subtype, these were determined according to EULAR criteria/ACR 2013.8 The modified Rodnan scale (mRSS) was used to assess the extent of disease; the prognosis of the patients was considered according to a consensus of EUSTAR experts.11 We inquired about clinical manifestations of cutaneous, pulmonary, gastrointestinal and renal involvement. Ischemic heart disease, peripheral vascular disease, hypertension, dyslipidemia, diabetes mellitus, chronic renal failure, osteoporosis (because of its high prevalence in the population) and toxic habits (which included smoking, alcohol, physical inactivity and obesity) were investigated.
Autoimmune serology was performed, consisting of antinuclear antibodies (ANA), anticentromere antibodies, anti-Scl-70, anti-RNP, anti-Sm, anti-SSA, anti-SSB, anti-Jo-1 and anti-dsDNA, in a subgroup of patients, using an electrochemiluminescent immunoassay method. Pulmonary artery pressure, pulmonary or tricuspid regurgitation and diastolic dysfunction were sought using echocardiography. The results of pulmonary function tests were divided into the following patterns: obstructive, restrictive and mixed. The presence of pulmonary fibrotic changes on the posteroanterior chest X-ray was investigated. HRCT was performed in 15 patients and the results were classified as normal, ground glass pattern, honeycomb pattern and others.
The focus of this study was to describe the findings in a series of patients with SS, so no treatment information was included.
Statistical AnalysisCategorical data are expressed in percentages, whereas continuous variables are presented using means±standard deviation (SD) when the distribution was normal, and using median and ranks when it was not.
Normality was assessed using the Shapiro–Wilk test. For the relationship between categorical variables we employed Fisher's exact test (FET). The relationship between a categorical variable and a continuous variable was determined by ANOVA or Kruskal–Wallis. All analyses were performed using α=0.05. The statistical package used was SPSS, v.20.
ResultsFour patients were excluded from the study for lack of information in the clinical records. 26 patients were included. The demographic characteristics of the patients are summarized in Table 1.
Demographic Characteristics of Patients With Systemic Sclerosis.
| n (%) | |
| Total | 26 (100) |
| Gender | 26 (100) |
| Female | 19 (73.1) |
| Average age at first symptom±SD (years) | 35.8±15.7 |
| <17 years | 4 (15.4 |
| 17–30 years | 6 (23.1) |
| 31–40 years | 4 (15.4 |
| 41–50 years | 5 (19.2) |
| 51–60 years | 6 (23.1) |
| >60 years | 1 (3.8) |
SD, standard deviation.
The prevalence of SS in patients in the Rheumatology department of the Hospital center (n=1.609) in the period March 2002–May 2013 was estimated at 1.62%. The prevalence by gender in this population was 1.35% for females (female patients in the period=1.406; female patients with SS=19) and 3.45% for male (male patients in the period=203, male patients with SS=7). The prevalence of diffuse SS in the department's patients (patients with diffuse SS=14) was 0.87%; 0.50% in females (female patients with diffuse SS=7) and 3.45% in males (male patients with diffuse SS=7). The prevalence of limited SS in the department's patients (limited SS=12) was 0.75%; there were no male patients with limited SS. So far, in the group of patients with SSc, both diffuse and limited, only one death occurred, equivalent to a mortality rate of 3.85%.
According to the latest government census, the population for which the rheumatology department serves as a unique center of excellence in public health is of 2805066 inhabitants, which can be divided into 1425725 males and 1379341 women. The prevalence of diffuse SS in this population is 5 and for limited SS is 4.3 per million, for a total prevalence of 9.3 per million inhabitants. The prevalence of these expressions of SS in males is 4.9 and 0 per million, respectively; in females it is 5.1 and 8.7 per million, respectively.
The age at diagnosis followed a normal distribution and the mean was 35.8years (median 39years), with a SD of 15.7. The age at which the first symptom related to SS appeared had normal distribution; the mean and SD are 32.6±15 years (median 36 years). 68% of patients were under the age of 40 at the time they had the first symptom related to SS; 73.1% of the population was diagnosed before age 50. In patients with diffuse disease, the mean age at diagnosis was 27±13.3 years (median 24 years), markedly lower than the mean age found in patients with limited SS, 46.1±12 years (median 46.5 years); the average age at onset preceded by 3 years the average age of diagnosis.
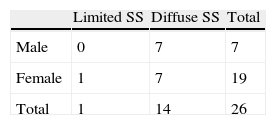
The majority of patients were female (73.1% [female:male ratio of 2.7:1]). All patients with the limited form of the disease were female; in the group of patients with diffuse SS, however, 50% of patients were male. When considering patients by age group, we found that, in patients of pediatric age and juveniles (n=4), the female: male ratio was 1:1. In the other age groups (17–30 years, 31–40 years, 41–50 years and >50 years) the majority of patients were female (66.7, 75, 80, and 83.3% respectively). There was a statistically significant relationship between subtype of scleroderma and gender (Table 2), (FET exact P = to .006).
Limited SS was found in 12 patients (43%), while the remaining patients (n=14) had diffuse SS. At the time that this study was performed, the average time of evolution (calculated using the age at onset of symptoms) was 6.8±4.3 years.
The extent of skin disease was determined using the mRSS which did not follow a normal distribution (Shapiro–Wilk P=.003). In patients with limited SS, the median was 14, with a range of 15 (minimum=6). When considering the division suggested by the EUSTAR consensus of experts in Paris, 2009, in this group of patients we found that 5.3% of patients have a high probability of remission (mRSS≤10); the rest have a 50% likelihood of improvement in symptoms (mRSS: 11–35). In patients with diffuse SS, median mRSS was 23, with a range of 39 (minimum value=6), with a minimum of 6; although 21.4% of patients had mRSS≤10 64.3% were found to have values in the range of 11–35. There was no statistically significant association between SS subtype and mRSS (H [1]=2.600; P=.107, mean range for limited SS=10.68, mean range for diffuse SS=15.57).
In both subtypes of SS, the process of induration was more severe in the fingers of the upper extremities, bilaterally, as evidenced by a median of 3 in the value of mRSS at this location. In both hands, the degree of general affection was moderate to severe, with values 2 and 3 for the median and mode of mRSS, respectively; however, in most patients, the left tended to be worse than its right counterparts regarding induration. In both arms, the median mRSS was 1.5 while the mode was 2, pointing to a mild-moderate condition in this location; both arms were equally affected.
In general, in the diffuse subtype of SS, the face and both feet were affected with mild (median and mode 1) induration, with no cases of moderate and severe disease. In the limited form of SS, facial skin involvement was rare (2 cases showed mild and 2 moderate induration), but the mild involvement of both feet was (median and mode 1) the norm.
The delay from the time of first symptom to the time of diagnosis had a mean of 2.6±2.7 years. Most patients (60%) were diagnosed in the year after disease began or the next; 5 patients took more than 5 years to be diagnosed after the first symptom. There was no relationship between the first symptom and diagnosis, and patient SS subtype (H [1]=0.480, P=.488; mean range for limited SS=14.14, mean range for diffuse SS=12.11).
Clinical ManifestationsThe most common clinical sign found was Raynaud's phenomenon; it was found in all patients with diffuse SS, but only in 54.5% of patients with the limited form. Localized calcinosis was found in 2 patients with diffuse disease; in patients with limited disease, 2 cases of localized calcinosis occurred, one case of diffuse calcinosis and another in a patient with visceral affection. In patients with diffuse SS, digital ulcers were found occasionally in 33.3% of cases and frequently in 13.3%; in patients with limited scleroderma, digital ulcers occurred frequently in 2 patients. The phenomenon of pitting was found in 46.7% of the patients with diffuse SS, but not in patients with limited SS.
Regarding respiratory symptoms, 66.7% of the patients with diffuse SS reported no dyspnea; of the 6 patients with dyspnea, 3 had dyspnea on moderate exertion. In patients with limited SS, one patient had great efforts dyspnea, while 2 had dyspnea on slight exertion. Cough was low in frequency, being present in only one patient with diffuse SS and in 2 patients with limited SS.
The most common clinical manifestation was gastrointestinal reflux, but appeared more frequently in patients with limited SS (54.5%) than in diffuse SSc patients (46.7%). Dysphagia was present in 26.7% of patients with diffuse SS, but only in 2 patients with limited SS. Clinical signs of malabsorption were found in a patient with diffuse disease and 2 patients with the limited form; constipation followed a similar distribution.
Renal manifestations were rare. No casts or azotemia was found. Three patients had albuminuria, and 2 with diffuse scleroderma. Erythrocyturia was found in a patient with diffuse disease. No patient had scleroderma renal crisis.
Fig. 2 summarizes the frequency of clinical manifestations in the patients with diffuse and limited SS.
Comorbidities and Toxic Habits are presented in this graph; the most frequent were Raynaud")
Additionally, the presence of comorbidities and toxic habits in this group of patients was studied. Consistently with the age range (73.1% of the population is under 50 years old) and the prevalence of female gender, the number of comorbidities was low. Fig. 3 summarizes the findings.
 was low, probably due to the fact that it was a predominantly young population. The most common comorbidity was hypertension. SS, systemic sclerosis.")
The comorbidity that was found more frequently in patients with both subtypes of SS was arterial hypertension: 26.7% and 45.5% of cases of diffuse and limited SS presented it, respectively. Dyslipidemia was found in 2 patients, one with diffuse SS and one with limited SS. Diabetes mellitus type 2 was found in 2 patients, one with diffuse SS and one with limited SS; there were no cases of obesity.
Cardiovascular disorders were observed in patients with limited SS: 2 had ischemic heart disease, and 3, peripheral vascular disease. There were no patients with diffuse SSc and cardiovascular disease.
Osteoporosis was found in a patient with diffuse SS and 3 patients with limited SS. Chronic kidney disease was demonstrated in a patient with limited SS.
The patients studied reported low risk lifestyles. The degree of physical inactivity in the population was low: only 4 patients reported sedentary life styles, 2 in each subtype of the disease. Regular use of tobacco was only seen in patients with limited SS. None of the patients reported regular intake of alcohol.
Complementary StudiesAs part of the study protocol, all the patients included in the study were evaluated using echocardiography. 2 cases of pulmonary arterial hypertension, a case of diastolic dysfunction and a case of pericarditis (none of these was the same patient) were found in the subgroup of patients with diffuse SS. The ejection fraction of the left ventricle was above 60% in all patients. 2 cases with pulmonary arterial hypertension and 3 with diastolic dysfunction were found in the subgroup of patients with limited SS; 2 patients showed isolated tricuspid regurgitation. There were no cases of pericarditis. Similar to the patients with diffuse mode, the ejection fraction of the left ventricle was above 60% in all patients. No statistically significant association between the presence of abnormalities on echocardiography and subtype of SS was observed (FET exact P=.680) or value of the mRSS (F[1,22]=0.239; P=.630).
The pulmonary function tests were normal in 6 patients (23.1%), 4 of these with limited scleroderma. Of the patients with diffuse SS, 60% had a restrictive pattern; there was one case with isolated obstructive pattern and a case with mixed pattern. In this group of patients, the mean values±SD for FVC, FEV1 and FVC/FEV1 were 69.5%±19.1%; 73.9% 22.7±107.4 and 16.6%, respectively. In the patients with limited SS it was found that 36.4% had normal spirometry, 36.4% had a restrictive pattern, 9.1% an obstructive pattern and 9.1% a mixed pattern. Mean±SD values for FVC, FEV1 and FVC/FEV1 in this subgroup of patients were 76.6%±24.4%; 84.1% 31.7±125.1 and 57.3%. There was no statistically significant association between the subtype of scleroderma and the development of a restrictive pattern (FET exact P=.408).
The posteroanterior chest X-ray images were suggestive of fibrotic changes in the lungs of 80% of the population with diffuse SS; in patients with limited disease, this finding was found in 54.5% of patients. For economic reasons, HRCT was performed only in 14 patients. Patients with diffuse SS on HRCT were 10, of which only one had a normal HRCT. Four patients (26.7%) had a standard “honeycomb” pattern; there were no patients with frosted glass pattern. In 5 patients there were other findings (bronchiectasis, pachypleuritis, increased pulmonary insterstitial images, emphysema and atelectasis). Of the 4 patients with limited SS in whom HRCT was performed, only one had no evidence of lung disease. A “honeycomb” pattern was found in one patient and a frosted glass pattern in another; other findings (bronchiectasis, pachypleuritis, increased pulmonary interstitium, emphysema and atelectasis) were found in 3 patients.
Until last year, there was no videocapillaroscopy equipment in the country and it currently remains a high cost study, not covered by social security; as a result, only 7 patients have videocapillaroscopy. Three patients with diffuse SS had videocapillaroscopy performed; one patient had no pathologic data, another had an active pattern and the third pattern had a late pattern. In the group of patients with limited SS, 2 had an active pattern, one an early pattern and one a late pattern.
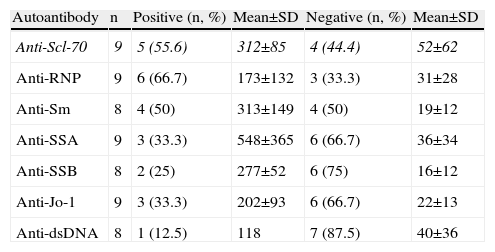
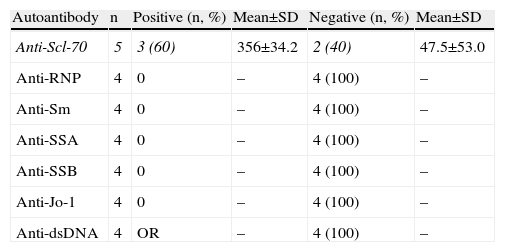
Autoimmune SerologyAn “autoimmune panel” composed of ANA, anticentromere, anti-Scl-70, anti-RNP, anti-Sm, anti-SSA, anti-SSB, anti-Jo-1 and anti-dsDNA antibodies was offered at a price subsidized by private institutions to help low-income population; anti-PM-Scl are not available in the country. Thirteen patients agreed to have autoimmune serology tests performed. Negative serology was found in one patient, who had limited SS. Tables 3 and 4 give details about the presence of each of these autoantibodies.
Basic Statistics Regarding Immune Serology in Patients With Diffuse Systemic Sclerosis.
| Autoantibody | n | Positive (n, %) | Mean±SD | Negative (n, %) | Mean±SD |
| Anti-Scl-70 | 9 | 5 (55.6) | 312±85 | 4 (44.4) | 52±62 |
| Anti-RNP | 9 | 6 (66.7) | 173±132 | 3 (33.3) | 31±28 |
| Anti-Sm | 8 | 4 (50) | 313±149 | 4 (50) | 19±12 |
| Anti-SSA | 9 | 3 (33.3) | 548±365 | 6 (66.7) | 36±34 |
| Anti-SSB | 8 | 2 (25) | 277±52 | 6 (75) | 16±12 |
| Anti-Jo-1 | 9 | 3 (33.3) | 202±93 | 6 (66.7) | 22±13 |
| Anti-dsDNA | 8 | 1 (12.5) | 118 | 7 (87.5) | 40±36 |
SD, standard deviation.
Italics are used to highlight autoantibodies of particular importance in systemic sclerosis.
Basic Statistics Regarding Immune Serology in Patients With Limited Systemic Sclerosis.
| Autoantibody | n | Positive (n, %) | Mean±SD | Negative (n, %) | Mean±SD |
| Anti-Scl-70 | 5 | 3 (60) | 356±34.2 | 2 (40) | 47.5±53.0 |
| Anti-RNP | 4 | 0 | – | 4 (100) | – |
| Anti-Sm | 4 | 0 | – | 4 (100) | – |
| Anti-SSA | 4 | 0 | – | 4 (100) | – |
| Anti-SSB | 4 | 0 | – | 4 (100) | – |
| Anti-Jo-1 | 4 | 0 | – | 4 (100) | – |
| Anti-dsDNA | 4 | OR | – | 4 (100) | – |
SD: standard deviation.
Italics are used to highlight autoantibodies of particular importance in systemic sclerosis.
ANA titers were investigated in 15 patients, 5 with limited SS and 11 with diffuse SS. All patients with diffuse disease had presence of ANA. In this group of patients there was a similar distribution between nucleolar (27.3%), speckled (45.5%) and homogeneous patterns (27.3%); there were no patients with anticentromere pattern. Four of the 5 patients with limited SS had ANA. In 2 cases, a homogeneous pattern was found; the other 2 patients had anticentromere mottled patterns.
DiscussionThe prevalence of SS for the general population was estimated to be 9.3 patients per million. This is significantly lower than the international average12–15 and, interestingly, is close to figures reported for12 Croatian and Dutch populations. The authors acknowledge the methodological limitations of this study used to determine the exact prevalence, but understand its value in a population and geographical area for which there are no published data. In the department's patients, during the period 2002–2013, it was found that the prevalence of SS in males is much higher than the prevalence in females; this is probably due to the low number of male patients referred to the department.
The mean age at diagnosis was lower than that reported in the literature,12,13,15 which shows an earlier onset in this population. A younger age was also reported in a subgroup of black patients, as compared with13 patients of white descent. It is likely that this early onset is related to the high proportion of patients with diffuse SS, which tends to occur at younger ages.
In this population, SS is associated with women, with a female:male ratio similar to the average value published by EULAR.12 Additionally, the influence of gender seems to be lower in this population than that reported in Spanish13,16 US or Canadian populations4 and a review of the literature.17 Moreover, it differs significantly from that reported for Buenos Aires, Argentina,14 which could be the result of a very different racial composition: the Dominican Republic has a strong presence of African and Taino genes (an American Indian population considered extinct until recently)7 while Argentina is known for its great waves of European immigration, which minimized indigenous, black, Asian or mixed18 races. In addition, it is noteworthy that half of the patients (50%) with diffuse SS were male and there is a statistically significant relationship (P=.006) between gender and subtype of scleroderma. This is consistent with that reported by the registry of the Spanish Network for Systemic Sclerosis, which also found greater male involvement in patients with the diffuse form of the disease,19 but differs from that found in a Chinese Han population.20
Diffuse SS is most frequently found than the limited form, but did not influence the time interval between the first symptom and diagnostics, although it has been reported that patients with diffuse disease are characterized by a shorter time to diagnosis.12 Additionally, the subtype of SS was not found statistically associated (P=.107) with the degree of skin condition, as measured by the value of mRSS. The location of the induration was similar in both subtypes of SS, although conditions such as pitting and digital ulcers were more common in diffuse SS. The respiratory system was found more frequently affected in patients with diffuse SS but was more severe when found in limited SS. Renal involvement was rare and always occurred in association with diffuse SS.
Although Raynaud’ phenomenon was present in all the patients with diffuse and more than half of patients with the limited form, and while reflux was reported in about half of patients in each subtype, most of the population has no clinical symptoms of severe organic disease. It is the opinion of the authors that the above is a result of early diagnosis in most patients, and not disease with benign course. This notion is supported by the fact that, although the average time of evolution is low, there is evidence of pulmonary disease in approximately 80% of patients and evidence of restrictive pattern in half of the population (heart disease and/or kidney affection were scarce).
Autoimmune serology for all patients was conducted, which introduces the possibility of bias, then, due to economic constraints, being generally conducted in patients with more severe disease. According to those reported in the literature,13 two patients had positive ANA, but one had limited SS. Anti-Scl-70 is found in most of the patients with the diffuse form and even in patients with limited SS consistent with clinical findings; additionally, anticentromere antibodies were found in patients with clinical findings of diffuse SS. Interestingly, in 2 cases there was coexistence of anti-Scl-70 and anticentromere. Similar findings have been described in other Latino populations, being rare in Caucasian or African American populations.21
Other autoantibodies were found in 3 patients with diffuse SS; although it is usually found associated with other diseases, and has also been described in patients with SS.21,22 The authors do not know the cause of these findings, but, in light of the literature,21 we speculate that the increased expression of anti-Scl-70 and anticentromere, the coexistence of these antibodies and the presence of other ANA in patients who meet criteria for SS (not mixed connective tissue disease, overlap syndrome and undifferentiated connective tissue disease) may result from the study population having a highly heterogeneous genetic load.
Interestingly, most of the patients were diagnosed in the first year after the onset of symptoms, which serves as encouragement to continue formal education of rheumatic diseases to medical undergraduates and primary care physicians, the primary source of medical referral in this population.
In summary, the prevalence of SS in the Dominican population is less than that reported internationally. The age of onset of the disease in the Dominican population appears to be lower than that reported in the literature. A different pattern of autoantibodies is observed in this population.
Ethical ResponsibilitiesProtection of people and animalsThe authors declare that this research has not performed experiments on humans or animals.
Data confidentialityThe authors declare that they have followed the protocols of their workplace on the publication of data from patients and all the patients included in the study have received sufficient information and gave written informed consent to participate in the study.
Right to privacy and informed consentThe authors have obtained informed consent from patients and/or subjects referred to in the article. This document is in the possession of the corresponding author.
Conflict of InterestThe authors state that there are no conflicts of interest.
Please cite this article as: Gottschalk P, Vásquez R, López PD, Then J, Tineo C, Loyo E. Esclerodermia en el Caribe: características en una serie de casos dominicana. Reumatol Clin. 2014;10:373–379.


