





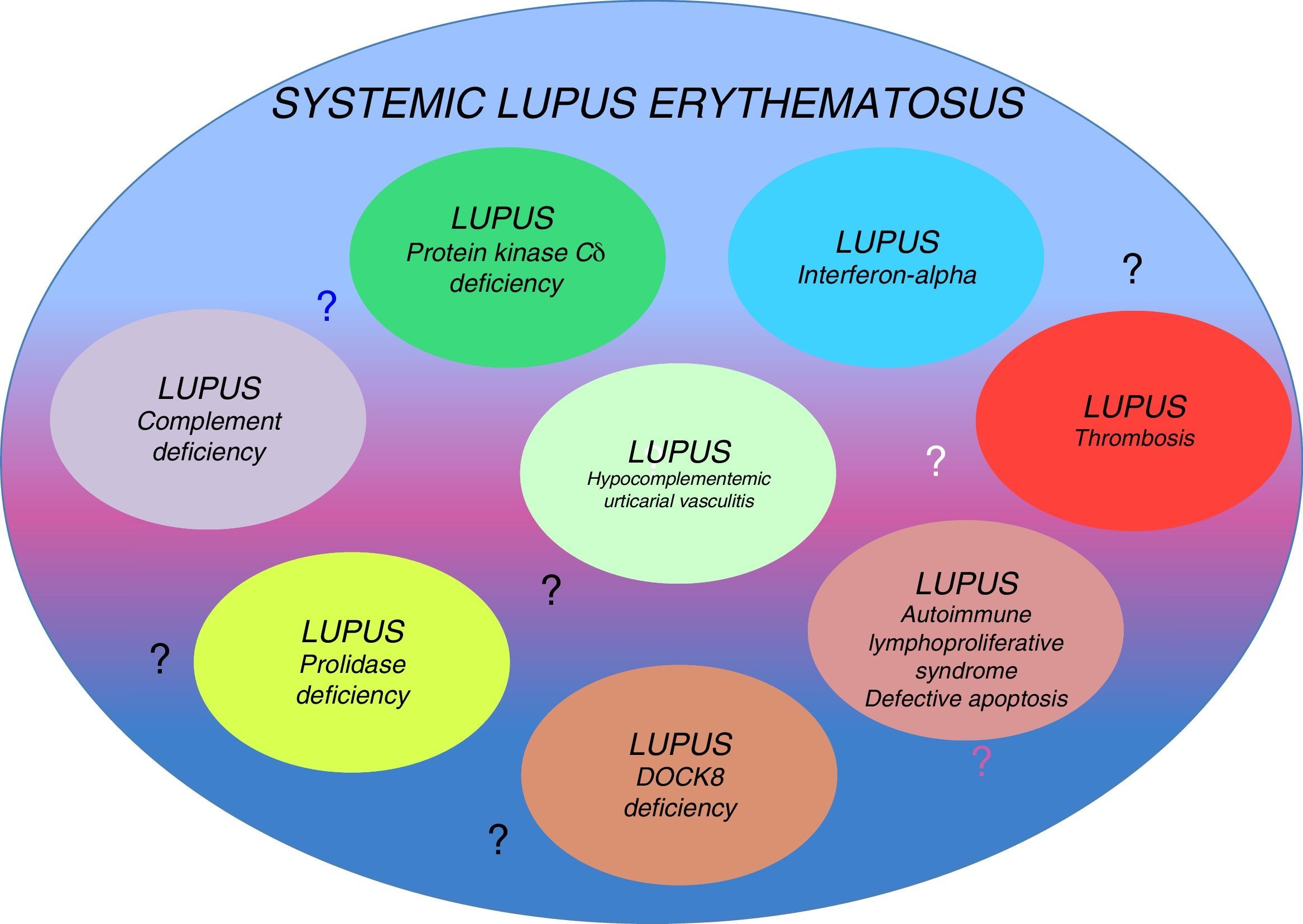
Systemic lupus erythematosus (SLE) is a multisystemic disease with a variety of clinical presentations. Monogenic predisposing conditions to the development of this disease have been described. As examples, an impaired expression of interferon-α regulated genes or complement deficiencies have been reported in patients with SLE, with particular clinical presentations. Those defects present particular presentations and a different severity, making an argument that lupus is not a single disease but many. Treatment could be individualized depending on the underlying defect generating the subtype of the disease.
El lupus eritematoso sistémico (LES) es una enfermedad multisistémica poseedora de una gran variedad de presentaciones clínicas. Se han descrito enfermedades monogénicas que predisponen la aparición de LES. Como ejemplos tenemos a los defectos en los genes reguladores de la expresión de interferón alfa o a nivel del complemento, que presentan comportamientos clínicos particulares. Estos defectos presentan una presentación y severidad distintas, por lo que se puede argumentar que el lupus no es una sola enfermedad sino varias. El tratamiento se podría individualizar dependiendo del defecto subyacente que genere el subtipo de lupus.
Systemic lupus erythematosus (SLE) is a multisystem disease with a wide variety of clinical presentations, caused by the production of autoantibodies, complement activation and immune complex deposition.1 Research on SLE is incredibly intense and, since 1946, around 56000 manuscripts touching on this subject have been published.
The onset of between 20% and 30% of the cases of lupus occurred during childhood. There are differences with respect to adult-onset lupus in that the female-to-male ratio is less marked in children (4:1, in contrast to 9:1 in adults) and the disease is more serious.2,3 Webb et al.4 reported a more severe phenotype of SLE in patients in whom onset took place at a pediatric age (pSLE), with a higher incidence of proteinuria, malar rash, anti-double stranded DNA (anti-dsDNA), hemolytic anemia, arthritis and leukopenia in comparison with adult-onset SLE. This where we begin to observe differences in the clinical picture of what we now know as lupus. Another marked difference in childhood-onset lupus is its strong association with primary immunodeficiencies (PID).5–7 In this respect, Liphaus et al.8 published an interesting study in which they analyzed 72 patients with pSLE, searching intentionally for possible cases of PID; they found a deficiency in 16 patients (22%). Three patients had C2 deficiency, another 3 had C4 deficiency, 2 had C1q deficiency, 4 had IgG2 deficiency, 3 had IgA deficiency, 3 had IgM deficiency and 1 had a simultaneous C2, IgA and C4 deficiency. It is noteworthy that, in this group of patients, the disease activity index was much higher compared to the rest of the patients. Thus, the authors suggest that in cases of severe pSLE, it is necessary to look for an underlying PID.
Genetic factors that have a fundamental influence on the development of this disease have been identified. Monozygotic twins have a 20%–40% higher risk of developing SLE, whereas in heterozygous individuals, the risk is only 2%–5% higher, suggesting the importance of genetic defects as determining factors of this disease, even if there is no associated inheritance pattern. Although the inheritance pattern is complex, they could be different conditions classified as a single disease. The existence of several genetic origins would explain the variety of phenotypes encountered in the clinical disease (that is probably the origin of reports of cases described as lupus-like).7,9 Occasionally, SLE has been associated with single-gene defects with well-defined clinical behaviors (Table 1). The relationship of genetic defects with the immune system and the development of SLE is a subject that is beginning to be discovered.9 In this review, our purpose is to provide arguments to support the concept of the existence of several different diseases (lupus-like diseases or subtypes of lupus) encompassed in what we know today as SLE (Fig. 1)
Single-gene Defects Associated With Systemic Lupus Erythematosus.
| Mutation | Mechanism | Clinical manifestations |
|---|---|---|
| Complement deficiencies (C1q, C1r, C1s, C2, C4) | Chronic infections Impaired clearance of apoptotic cells and immune complexes | Recurrent infections Early onset Prominent cutaneous manifestations Absence of anti-dsDNA antibodies |
| PKCδ deficiency | Excessive B-cell proliferation (apoptosis) | Hepatosplenomegaly Lymphadenopathy EBV or CMV infection |
| Prolidase deficiency | Alteration in C1q | Recurrent infections Elevated IgE Cutaneous manifestations (telangiectasias) Facial dysmorphia |
| Interferonopathy (TREX1, IFIH1, ADAR) | Increase in IFNα | Lupus pernio Basal ganglion calcification |
| RASopathy (RAS/MAPK pathway) | Leukocyte proliferation Defective apoptosis | Increased rate of pericarditis Little skin involvement Splenomegaly, lymphadenopathy Association with Noonan syndrome Risk of malignant transformation |
| Mutation in DNASE1L3 | Alteration in DNA removal | Urticaria, cutaneous vasculitis, Recurrent abdominal pain, uveitis, episcleritis, glomerulonephritis with anti-C1q antibodies |
CMV, cytomegalovirus; dsDNA, double-stranded DNA; EBV, Epstein–Barr virus; PKC, protein kinase C.
We know that patients with deficiencies in the initial phases of the complement pathways (C1q, C2 and C4) have a greater predisposition for developing autoimmune diseases. In fact, C1q deficiency is considered to be the genetic factor with the strongest risk and fraction C2 deficiency to be that most frequently associated with SLE.10–12 For a clinical datum, we have the fact that lupus generally has an early onset and there is no predominance of one gender over the other. Moreover, it is associated with the presence of recurrent infections. Why do patients with complement deficiencies develop more severe autoimmunity? There are several factors that could act alone or together: (1) as a consequence of the constant immune activation by a chronic viral infection; (2) an association with human leukocyte antigen (HLA), as the complement components C2, C4 and factor B are in the major histocompatibility complex; (3) the importance of complement in immune tolerance; (4) the role of complement in the solubilization of the immune complexes by reducing their size and increasing their clearance from the circulation; the immune complexes bound to C3b bind to the C3b receptor located in erythrocytes (CD35), which, in this way, are transported to the Kupffer cells in the liver and their renal deposition is prevented; and (5) the role of complement, particularly C1q, in the clearance of apoptotic cells, which, being impaired, permits a greater exposition to autoantigens.13–17
There are differences between the rheumatic diseases of patients with complement deficiency when compared with those in whom this deficiency is absent. One example is the case of patients with C2 deficiency, who are more likely than others to have photosensitivity dermatitis, low positivity for anti-dsDNA antibodies and high anti-Ro titers.18–20 On the other hand, patients with C1q deficiency have a very early onset, prominent cutaneous manifestations and substantial damage to target organs, with cerebritis and nephritis.21,22
There are multiple mutations that occur on each of the 3 genes responsible for C1q deficiency, which are found in the long arm of chromosome 1.21
Approximately 38 families have been reported to have confirmed C1q deficiency, and it is estimated that 90% develop SLE.21 In a study of 41 patients, the majority had rash (98%), and glomerulonephritis was also common (39%). An interesting finding is that tests for anti-dsDNA antibodies are usually negative.23
The case reported by Arkwright et al.24 is particularly relevant: a 17-year-old boy with a history of parental consanguinity and the deaths of his brothers due to infections, in whom C1q deficiency and SLE were detected. He underwent hematopoietic stem cell transplantation, which cured his SLE. The majority of the complement factors are proteins that are synthesized in the liver. For this reason, patients with this type of PID do not usually undergo hematopoietic stem cell transplantation; however, the explanation for the performance of transplantation in the aforementioned patient is the monocyte production of C1q, with the resulting resolution of the disease.24
C2 deficiency is the most common homozygous deficiency, with a prevalence of 1 per 20000 population. In all, 10%–20% of the cases have been associated with SLE. The main manifestations of patients with C2 deficiency are infections; approximately 50% of the patients are susceptible to infectious processes, especially sepsis, meningitis, arthritis and osteomyelitis caused by encapsulated bacteria (Hemophilus influenzae, pneumococci, meningococci). When they develop autoimmunity, the system most affected is the cardiovascular system, and tests for anticardiolipin antibodies are frequently positive. The cause of death is early atherosclerosis, as well as infectious processes. Other deficiencies, like those of C4 and properdin, have also been associated with the development of SLE. C3 deficiency is also associated with recurrent infections, but can also produce signs of lupus, such as fever, rash, glomerulonephritis and arthritis, in up to 28% of the patients. It has been reported that the use of fresh frozen plasma could be a therapeutic alternative, as could rituximab.25
Systemic Lupus Erythematosus: An Interferonopathy?The behavior of type I interferons (IFN) is highly variable and depends on the diseases with which they are related. For example, while in patients with multiple sclerosis and arthritis they have an inhibitory effect on the Th17 response and, thus, on the disease activity in SLE, they are capable of inducing disease activity through different mechanisms.26–31 Type I IFN comprise around 13 molecules, including IFNα, IFNβ, IFN¿, IFNκ and IFNω, and the genes that encode them are located on chromosome 9. Interferons have the capacity to activate the innate immune response through Toll-like receptors 3, 7, 8 and 9. They are mainly produced in the nucleated cells, plasmocytoid dendritic cells and monocytes. It has been found that, in SLE patients, these cytokines are capable of inducing the production of immune complexes directed against nucleic acids, this through IFN regulatory factors.28
The effects of IFNα in a healthy population involve the maturation of dendritic cells, activation of natural killer (NK) cells, increases in the Th1 immune response, in CD8+ T lymphocytes, in memory cells and in B-lymphocyte stimulator (Blys/BAFF) and an improvement in immunoglobulin response. In patients with SLE, while they increase the levels of Blys, on the other hand, they reduce the regulatory cells (CD4-FoxP3).32
A clinical example that involves a well-established genetic defect, and includes the association of SLE and IFN, is Aicardi-Goutières syndrome (AGS), first described in 1984 by Jean Aicardi and Françoise Goutières, who detected patients with calcifying vasculitis, leukodystrophy and progressive encephalopathy in the absence of infection caused by the TORCH syndrome (Toxoplasma gondii, Others [human immunodeficiency virus, Treponema pallidum, etc.], Rubella, Cytomegalovirus [CMV] and Herpes virus). In 1988, Pierre Lebon detected high IFNα levels and lymphocytosis in the cerebrospinal fluid of patients with this syndrome; IFNα is even considered a prenatal marker that can lead to the suspicion of this disease.33,34
In 2006, Yannick Crow and Andrew Jackson and their collaborators identified mutations in 4 different genes: TREX1, RNASEH2B, RNASEH2C and RNASEH2A; subsequently, a fifth mutation, referred to as SAMHD1, was discovered. These 5 defects are estimated to account for 90% of the mutations known to cause AGS.35 More recently, other genes involved have been identified: SAMHD1, ADAR and IFIH1.
In a review of 26 patients diagnosed with AGS, Ramantani et al.28 report a high prevalence (60%) of manifestations associated with SLE, such as thrombocytopenia, leukopenia, positive test for antinuclear antibodies (ANA), erythematous lesions, lupus pernio, oral ulcers and arthritis. This fact leads to the suspicion that the same genetic defect that predisposes to AGS has an impact on the immune system that involves a risk factor for the development of lupus. The common denominator between AGS and SLE known at the present time is the series of metabolic changes induced by the IFN, specifically IFNα. The correlation between high levels of this cytokine and the activity of lupus is well-established. There are reports of patients who have both AGS and SLE,32,36 in addition to heterozygous mutations in TREX1 in chilblain lupus.37–39 The mutation of TREX1 has been related to an autoimmune spectrum that includes AGS, familial lupus pernio, retinal vasculopathy with cerebral leukodystrophy and SLE itself. The TREX1 gene encodes a DNA exonuclease and its mutation produces a degradation of dysfunctional DNA.40–43 The same occurs with mutations in IFIH1, with a broad spectrum in relation to the phenotypes encountered. This defect was found in a 16-year-old patient with early onset SLE that was refractory to treatment.44 Moreover, when the whole exome was sequenced, the pathogenic mutation was detected in another SLE patient, a 4-year-old girl with cerebral involvement.40 In a series in which the molecular defect was characterized in a large group of patients with AGS, only 4 had SLE (2ADAR, 2IFIH1), indicating that the frequency is low. Some of the patients had oral ulcers, progressive arthropathy or intracranial vasculopathy, evidence that the spectrum of inflammatory manifestations is extensive.45
Stimulator of interferon genes (STING) is a transmembrane protein that resides as a dimer in the endoplasmic reticulum of epithelial and endothelial cells, macrophages and dendritic cells. It is essential for the production of cytokines like type I IFN in response to pathogens related to double-stranded DNA. Mutations affecting STING have been related to some forms of SLE. Four members of a family had a systemic inflammatory syndrome with vasculopathy, pulmonary fibrosis and autoimmunity.46 The index case had fever, malar rash, lung disease and positive tests for ANA and anti-dsDNA antibodies. One current line of research is the evaluation of therapy aimed against IFNα in SLE (rontalizumab).47–49
RASopathiesSomatic gain-of-function mutations in members of the subfamily of RAS guanosine triphosphatases have been found in up to 30% of all human cancers. Variants of autoimmune lymphoproliferative syndrome have been reported to carry mutations in NRAS and KRAS, two members of this family.50–57 This entity has been referred to as RAS-associated autoimmune leukoproliferative disease, and the patients have splenomegaly, lymphadenopathy, autoimmune cytopenias and hypergammaglobulinemia, as well as recurrent infections. There is a breakdown of leukocyte homeostasis affecting apoptosis. RASopathies are a group of genetically-defined diseases that include Noonan syndrome (NS) and other related conditions. These syndrome are characterized by neurodevelopmental defects that result in mutations in genes encoding components or regulators of the RAS/MAPK pathway.57 There are cases of NS with single-gene defects associated with the development of SLE reported in the literature.52–56 Such is the case of a 13-year-old boy with a diagnosis of NS and hair loss, with a well-defined mutation in SHOC2. He had pericarditis, polyarthritis, massive lymphadenopathy, hepatosplenomegaly and positive tests for ANA and anti-dsDNA. He was treated with aspirin and hydroxychloroquine, which resulted in remission of all the symptoms except hepatosplenomegaly and lymphadenopathy.57 For this reason, Bader-Meunier suggests that some SLE patients could have a RASopathy.52,53
Protein Kinase-Cδ DeficiencyBelot et al.58 reported the cases of 3 patients, born to consanguineous parents, who met SLE criteria. In 1 of them, the manifestations were suggestive of autoimmune lymphoproliferative syndrome with hepatosplenomegaly and lymphadenopathy. All 3 patients presented with nephritis and positive tests for ANA and anti-dsDNA, and some had cutaneous, joint, neurological and hematological involvement. Protein kinase-Cδ (PKCδ) is a serine/threonine kinase involved in the control of cell proliferation and apoptosis, and its absence is associated with an excessive proliferation of B cells. We should point out that the patients did not have recurrent infections.59 On the other hand, Kuehn et al.59 report another case of PKCδ: a Mexican male from an endogamous community, who did have recurrent infections (otitis, sinusitis), in addition to chronic Epstein–Barr virus infection. The findings in this patient were indicative of autoimmune lymphoproliferative syndrome with elevated transaminase levels and hyperglobulinemia but, although he was not diagnosed with SLE, he had malar rash and positive tests for ANA and anti-Sm antibodies. Kiykim et al.60 report the case of a 3-year-old boy, the son of consanguineous parents, with early manifestations including intermittent fever, myositis, alopecia, photosensitivity, hypotonia, cervical lymphadenopathy and hepatosplenomegaly. A skin biopsy revealed lupus. The patient had CMV and decreased NK-cell function. The genetic study identified a novel mutation in PKCδ. According to the authors, the patient had a spectacular response to hydroxychloroquine, in combination with trimethoprim and intravenous immunoglobulin. Thus, in this subtype of SLE, there is a predominance of lymphoproliferation and, in some cases, susceptibility to infections.
Prolidase DeficiencyAnother genetic defect that can be a determining factor in SLE is prolidase deficiency, an extremely rare disease with an estimated incidence of 1 per million births. Some 70 cases have been reported in the literature. Its inheritance pattern is autosomal recessive and it is characterized by chronic skin lesions, splenomegaly, peculiar facies, mental retardation, multisystem involvement (esophageal, pulmonary), iminodipeptiduria and elevated immunoglobulin levels, especially IgE. Prolidase is a ubiquitously distributed dipeptide involved in the last stages of degradation of endogenous and dietary proteins.61–65 It is essential in collagen catabolism as it is involved in the hydrolysis of peptides containing proline or hydroxyproline. The hypotheses concerning the pathogenesis point to changes in connective tissue metabolism that produce angiopathy or a change in C1q, which contains high amounts of proline. There is documented evidence of an important role of prolidase in the maturation and activation of the type I IFN receptor. The susceptibility of these patients to recurrent infections (pneumonias, sinusitis, otitis and CMV and herpes virus infections) is attributed to the alteration in the expression of this receptor. The definitive diagnosis is based on the documentation of the mutation in the PEPD gene. The most frequently reported dermatological manifestations are recalcitrant skin ulcers on the legs, in addition to telangiectasias and photosensitivity.61 Onset can occur from birth to the age of 22 years. There are reports of patients with this defect with hypocomplementemia and positive tests for anti-dsDNA and anti-Sm antibody, which are characteristic of SLE. This diagnosis should be considered in patients with SLE and recurrent infections, facial dysmorphia, developmental delay, skin lesions and elevated IgE level.61–65
Lupus and HypogammaglobulinemiaPatients with SLE typically have hypergammaglobulinemia.66 Rankin and Isenberg67 have reported that up to 5% of the SLE patients have selective IgA deficiency. Patients with SLE can have concomitant antibody deficiency or develop it later on. It is important to determine whether there is renal loss of IgG secondary to nephrotic syndrome. On the other hand, there are a number of cases associated with the presence of common variable immunodeficiency. This disease is the most common of the symptomatic primary immunodeficiencies, and the diagnosis is based on the decrease in IgG levels, but in IgA and/or IgM levels as well (in contrast to the renal losses, in which only IgG decreases), with an alteration in specific antibody production of.68–71
Hypocomplementemic Urticarial VasculitisHypocomplementemic urticarial vasculitis is characterized by recurrent urticarial, cutaneous vasculitis, arthritis and glomerulonephritis. It is reported to be associated with SLE in more than 50% of the cases. Onset is usually in the third and fourth decades of life. The particular characteristics of this disease are uveitis, episcleritis, recurrent abdominal pain and severe glomerulonephritis with low C1q and anti-c1q antibody levels. There continues to be debate as to whether to consider this disease as a separate entity or a particular subtype of SLE. It is associated with mutations in DNASE1L3 (loss-of-function) in a family with 3 affected children.72 The protein encoded by DNASE1L3 is homologous to DNase-1 and behaves like an endonuclease, degrading DNA. This is responsible for removing DNA from nuclear antigens. Notably, the patients reported to have the mutation met criteria for SLE.73 Anti-C1q antibodies were found in all of them, whereas in SLE, they are present in only 30% to 35% of the patients. In our experience, intravenous immunoglobulin, in combination with the standard treatment, has been an effective therapeutic option.74 The whole conceptual controversy surrounding this disease is one more argument in support of the hypothesis of the existence of several subtypes of lupus.
Other ImmunodeficienciesDOCK8 deficiency is a determining factor in what is known as autosomal recessive hyper-IgE syndrome, susceptibility for viral, bacterial, fungal and protozoan infections, in addition to atopic diseases with elevated IgE levels.
DOCK8 is normally expressed in cells of the immune system and determines several effects on the lymphocytes. The loss of DOCK8 expression causes defects in T-cell proliferation and the production of antiviral cytokines, and a decrease in interleukin (IL)-17 (important in the defense against fungi). It also is determinant in an increase in the production of cytokines associated with the Th2 immune response, with eczema and eosinophilia. The patients show clinical signs of susceptibility to viral infections (especially cutaneous infections, herpes, verrucae and molluscum contagiosum) and autoimmunity (vasculitis, autoimmune hemolytic anemia).75–78
There are reports of 3 cases of SLE associated with hyper-IgE syndrome in pediatric populations.75–77 The first patient was an 11-year-old Asian boy who had been diagnosed with hyper-IgE syndrome at the age of 6 months. The second patient was a 5-year-old girl, the daughter of consanguineous parents, with a history of herpetic blepharitis, pyodermitis, eczema, recurrent pneumonia and purulent otitis media, with elevated IgE levels and DOCK8 deficiency. She developed arthritis, discoid lupus, purpuric lesions on the skin that was exposed to the sun, positive tests for ANA and anti-dsDNA antibodies and a skin biopsy consistent with chronic lupus erythematosus. She was treated with hydroxychloroquine and antibody prophylaxis and, at the time of the publication of the her case, stem cell transplantation was planned. Several years ago, we reported the case of a third patient, a 10-year-old Mexican girl with hyper-IgE syndrome, SLE and secondary antiphospholipid syndrome (cerebral infarction); the patient had recurrent infections.76,77
Patients with chronic granulomatous disease (CGD) and mothers who are carriers of the disease are predisposed to the development of several forms of lupus.79–81 One hypothesis to explain this phenomenon involves defective neutrophil apoptosis with production of autoantibodies.81 Chou et al.82 report a patient, the daughter of consanguineous parents, with chronic diarrhea and arthralgia, who developed SLE at the age of 8 years. She had hepatosplenomegaly, elevated transaminase levels, autoimmune hemolytic anemia, and positive tests for ANA and anti-dsDNA antibodies. Whole exome sequencing enabled the identification of a mutation in NCF2 of the NADPH oxidase complex (CGD).
Single-gene diseases predisposing to autoimmunity have recently been recognized. Mutations in RAG1 and RAG2 in humans result in a wide spectrum of phenotypes, from the absence of T cells and B cells with severe combined immunodeficiency to late autoimmune disease or granulomas. The autoimmune phenotype can vary widely, from cytopenias to destructive vasculitis. Loss of tolerance is due to altered lymphocyte receptor editing, and patients with RAG deficiency have been reported to produce a considerable variety of autoantibodies, including those characteristic of SLE.83 It is important that we point out that patients with this disease produce anti-IFN antibodies, and this is associated with severe viral infections. These same anti-IFN antibodies have been found in SLE patients.84 The case of a 44-year-old woman with mutations in RAG2 and SLE was recently described. She had polyarthritis, Raynaud's phenomenon, sicca symptoms, serositis, rash and nephritis, in addition to hypocomplementemia and positive tests for anti-Sm, anti-RNP, anti-Ro and anti-dsDNA antibodies.85 Curiously, not only did she have T-cell lymphopenia, but B-cell lymphopenia as well, and recurrent infections. There are also reports of SLE patients with mutations in FasL. Wu et al.86 described the case of one such patient and suggest that this entity be considered in patients with SLE and marked lymphadenopathy (in addition to the aforementioned conditions). We have observed a case of Mendelian susceptibility to mycobacterial disease (an IL-12 receptor beta chain defect), in which the patient also had tuberculous meningitis and SLE.87 This association is noteworthy because, contradictorily, in animal models, IL-23 receptor deficiency protects against the development of SLE.88 Although autoimmune phenomena are common in patients with Wiskott-Aldrich syndrome, only 1 case associated with SLE has been described.89
Polymorphisms in cytotoxic T lymphocyte antigen-4 (CTLA-4), a primordial regulatory protein of the immune response, have been reported in SLE patients.90 Loss of CTLA-4 is fatal in mice. On the other hand, there are families diagnosed with an immunodeficiency syndrome in which autoimmunity is produced by heterozygous mutations in CTLA-4.91,92 Although the patients were not diagnosed with lupus, they had severe autoimmunity, with cytopenias, enteropathy and granulomatous pulmonary infiltrate. They also had recurrent infections and hypogammaglobulinemia.91,92 Patients with CTLA-4 haploinsufficiency are expected to benefit from soluble CTLA-4 fusion proteins (abatacept and betacept), which inhibit immune activation. Abatacept has been shown to have some effect in SLE, although the findings are inconsistent.93 Thus, in the future, the documentation of the particular defect in each lupus patient could help to guide the treatment more coherently.
Finally, early-onset Evans syndrome (hemolytic anemia and thrombocytopenic purpura) with immunodeficiency and premature immunosenescence has been found to be related to tripeptidyl-peptidase II deficiency.94
ConclusionsThere are a number of differences between SLE and pSLE, mainly in terms of chronicity, severity and the association with PID, a circumstance that suggests the possibility that SLE and pSLE are different in many aspects.
It could be that early-onset SLE is not a single disease, but occasionally comprises a heterogenic group of different single-gene defects associated with susceptibility to infections and involving different organs.95
As the manifestations of PID continue to be described, the findings include an increasing number of hypomorphic mutations that may cause PID with a milder phenotype and onset in adulthood. It could be that the same occurs with the defects encountered in SLE (or that different novel mutations are to be found).96
The association between elevated IFN levels and changes in the Th17 immune response is an area under study because of its impact on the development of SLE and its important therapeutic implications.17,21 Knowing the type of SLE we are dealing with could help in the choice of a better guided treatment (intravenous immunoglobulin, anti-IFNα antibodies, abatacept, rituximab, hydroxychloroquine, antibiotic prophylaxis or hematopoietic stem cell transplantation) (Fig. 2).
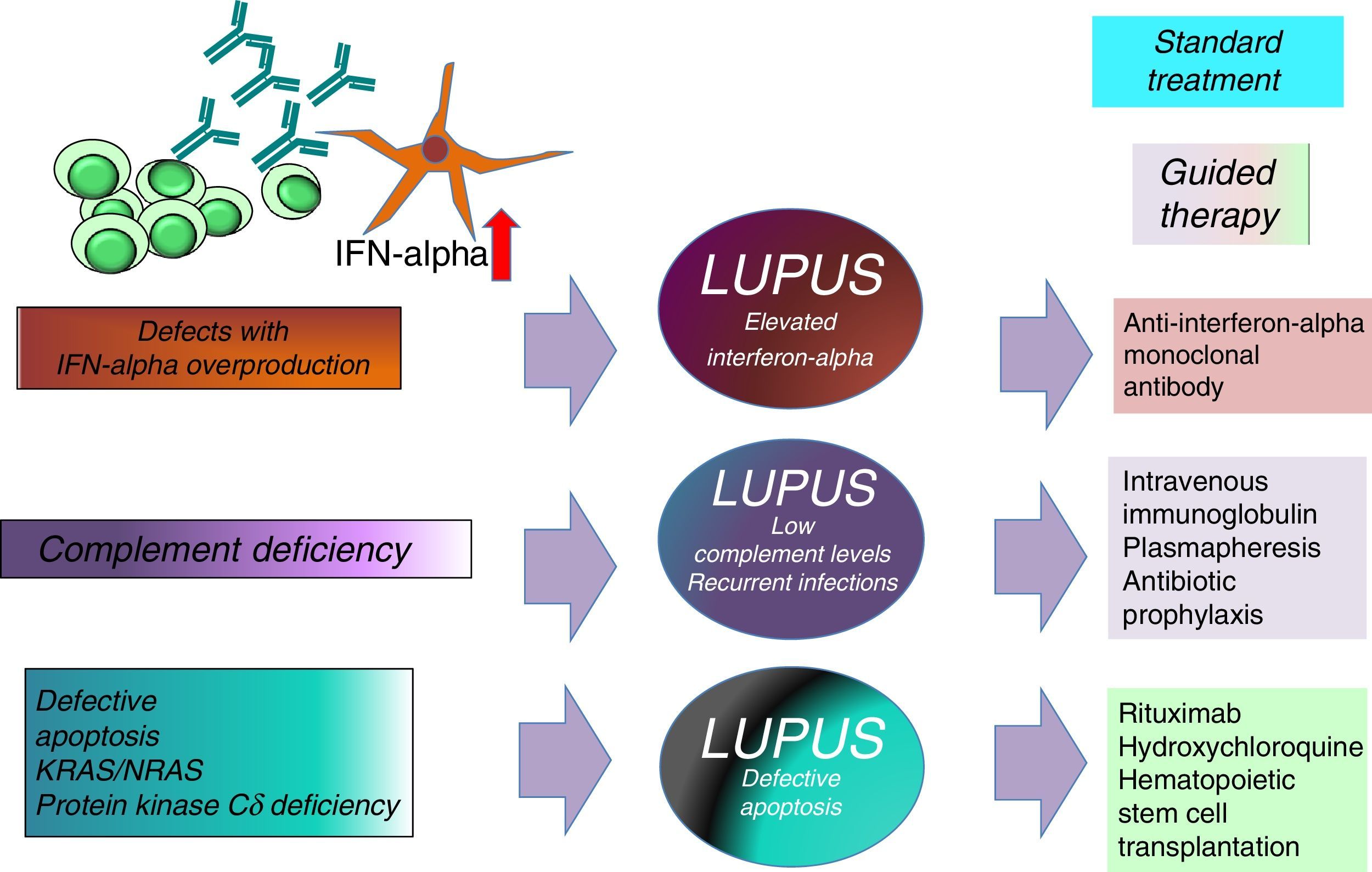
Finally, in any patient with severe pSLE associated with life-threatening or recurrent infections or infections caused by less common microorganisms (Pneumocystis jiroveci, tuberculosis, candidiasis, aspergillosis, coccidiodomycosis, CMV) associated with extremely low complement levels and/or parental consanguinity, the approach should be to rule out the presence of a PID that could have an effect on the disease course.23 Thus, in this respect, lupus is not a single disease, but several diseases with different etiologies, severities, onsets and treatments.
Ethical DisclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that no patient data appear in this article.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflicts of InterestThe authors declare they have no conflicts of interest.
Please cite this article as: Rivas-Larrauri F, Yamazaki-Nakashimada MA. Lupus eritematoso sistémico: ¿es una sola enfermedad? Reumatol Clin. 2016;12:274–281.


