





In 2015 the Spanish Society of Rheumatology (SER) published its position paper on biosimilar drugs. In this update, the SER, continues to manifest its unequivocal commitment to the sustainability of the health system of our country and is aligned with the measures that, without reducing quality of care, are aimed at ensuring its continuity. Since the publication of the previous position paper, the European Commission has authorised new biosimilar drugs, which provides an excellent opportunity to advance the efficiency of health care. In this new scenario of increased therapeutic offer of biologics, the SER considers it crucial to preserve the freedom of prescription of physicians who prescribe drugs based exclusively on the characteristics and individual circumstances of each patient, without forgetting the economic aspects thereof.
En el año 2015 la Sociedad Española de Reumatología (SER) publicó su posicionamiento sobre fármacos biosimilares. En esta actualización, la SER, sigue manifestando su compromiso inequívoco con la sostenibilidad del sistema sanitario de nuestro país y se alinea con las medidas que, sin reducir la calidad asistencial, estén encaminadas a asegurar su sostenibilidad. Desde la publicación del anterior posicionamiento la Comisión Europea ha autorizado la comercialización de nuevos fármacos biosimilares, lo que abre una excelente oportunidad de avanzar en la eficiencia de la atención sanitaria. En este nuevo escenario de incremento de la oferta terapéutica de biológicos, la SER considera imprescindible preservar la libertad de prescripción de los médicos que realizan la indicación de fármacos basándose exclusivamente en las características y circunstancias individuales de cada paciente, sin olvidar los aspectos económicos que se derivan de dicha actuación.
The introduction of biological drugs (BD) has transformed the treatment of chronic inflammatory diseases. Their efficacy and safety profile has made it possible for many patients who used to have reduced quality of life to undertake normal activities. The BD have a major economic impact, and their use has required an important effort by the Spanish National Health System.1
The authorisation of commercialisation by the European Commission following the approval by the European Medicines Agency (EMA) of drugs which are biosimilar (BSD) to the original BD has given rise to an opportunity for progress in the efficiency of healthcare. According to the EMA, a BSD is a BD that contains a version of the active component of an original biological product that has already been approved (the reference drug [RD]).2 To be approved, a BSD drug has to be directly compared with the original to show that the differences respecting the latter have no effect on its safety and efficacy.
The inclusion of BSD in normal clinical practice leads to new challenges, such as doctors’ freedom to prescribe when selecting which target to block with which product, the suitability of mass substitution for economic reasons, or exchangeability as decided by the patient and rheumatologist. These challenges also include the possibility of replacing one BSD with another due to exclusively economic criteria and without any formal evaluation by clinical trials (CT).
In 2015, the Spanish Society of Rheumatology (SER) published its opinion on the use of BSD for rheumatic diseases.3 The present paper updates this opinion in the light of new data which have appeared since the initial publication.
EMA normsThe EMA, which pioneered the development and establishment of the regulatory process for the authorisation of BSD, is in charge of evaluating requests for the approval of BD, including BSD, prior to their commercialisation in the European Union (EU). The EMA has published several scientific guides on BSD (Table 1). According to EU law on medicines, BSD have to undergo a centralised registration procedure coordinated by the EMA, and they are evaluated by the same experts who evaluate the RD. The Committee for Medical Products for Human Use (CHMP) issues a scientific opinion on approval, and finally the European Commission (EC) reaches a final decision on approval for commercialisation in the EU. Although this centralised procedure is valid in all EU countries, the EMA makes no recommendations about the interchangeability of RD and BSD, and each state has to decide on this question based on the available scientific information and their respective legal frameworks. The EMA specifies that any decision on interchangeability must involve both the prescriber and the patient.4
The main EMA guides on biosimilar drugs.
| Guideline on similar biological medicinal products | CHMP/437/04 Rev 1Published 29/10/2014 |
| Guideline on similar biological medicinal products containing biotechnology derived proteins as active substance: Non clinical and clinical issues | EMEA/CHMP/BMWP/42832/2005 Rev 1Published 09/01/2015 |
| Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues | EMA/CHMP/BWP/247713/2012Published 03/06/2014 |
| Guideline on similar biological medicinal products containing monoclonal antibodies: non-clinical and clinical issues | EMA/CHMP/BMWP/403543/2010Published 15/6/2012 |
| Guideline on immunogenicity assessment of biotechnology-derived medical products | EMEA/CHMP/BMWP/14327/2006 Rev.1Published 1/06/2017 |
| Immunogenicity assessment of monoclonal antibodies intended for in vivo clinical use | EMA/CHMP/BMWP/86289/2010Published 15/06/12 |
| Comparability of biotechnology-derived medicinal products after a change in the manufacturing process - non-clinical and clinical issues | EMEA/CHMP/BMWP/101695/2006 Published 19/07/2007 |
| ICH Topic Q 5 E Comparability of Biotechnological/Biological Products | CPMP/ICH/5721/03Published 01/06/2005 |
The use of BSD is controversial, especially in connection with their interchangeability and replacement, as although these concepts seem to be similar, from a technical-medical-legal standpoint they are different, and managers’ economically-based criteria may affect freedom of prescription.
In Spanish law, while awaiting specific regulation for BSD, the basic norm to be followed is contained in Law 29/2006, of 26 July, on guarantees and the rational use of medicines and medical products, which interprets substitution to be an act by means of which a pharmacist may, exceptionally and for specific reasons such as urgency or lack of supply, dispense a medicine other than the one prescribed by a doctor, replacing it with one having the same composition and pharmaceutical form, form of administration and dosage. This possibility does not apply to medicines which, due to their bioavailability characteristics and narrow therapeutic range, have been excluded by the Ministry of Health and Consumption. Thus the possibility of substitution by a pharmacist is restricted to identical products, and this is not the case for BD and their corresponding BSD. In Order SCO 2874-2007 of 28 September, the list of medicines which cannot be substituted was published. This includes the BD, and the Informative Note of the Spanish Agency for Medicines and Medical Products dated 24/4/2009 expressly mentions this.
Regarding interchangeability, article 89.5 of the Law on Guarantees (Legislative Royal Decree 1/2015, of 24 July) states that “in the case of BSD medicines, the norms in force will be obeyed according to the specific regulation on substitution and interchangeability”, indicating that the BSD are governed by a different legal regime, even though their specific regulation is not defined. There is controversy regarding whether this applies solely to pharmacies and not to hospitals. It should be remembered that currently BSD, such as those for insulin, which are commercialised in pharmacies, so that it would be a contradiction in terms if substitution were prohibited in pharmacies and permitted in hospitals.
The government of Spain, in reply to a parliamentary question, admitted that “biosimilar medicines are BD and as such cannot be substituted or interchanged in dispensing without the knowledge of the prescribing doctor” (BOCG, series D No. 472, 5 June 2014, page 345).
According to Spanish law, it is clear that BD cannot be substituted without the approval of the prescriber, so that any change between BD will have to be made on the initiative of or in agreement with the prescriber, so that legally this will not be a substitution but rather an exchange.
Apart from legal considerations, there is full agreement between scientific societies regarding the need for doctors to take part in the decision of interchangeability, expressing their respective opinions. Only the Spanish Hospital Pharmacy Society clarifies one point, attributing Pharmacy and Therapeutic Commissions with the capacity to decide on interchangeability within hospitals, given that these commissions contain prescriber representatives. This possibility of group decision-making is not acceptable, given that the norm which governs the working of the said commissions (Royal Decree 521/1987, of 15 April) does not confer this competence on them, and the responsibility for treatment is personal and does not correspond to a group but rather the doctor in question, who is solely responsible for prescribing, according to article 79 of the Law of Guarantees.
Information for the patient and freedom of prescriptionThe doctor-patient relationship is based on trust, communication and patient autonomy, so that the latter has to be informed about the treatment they are offered, and they have to agree to this. This demand is social and legal, and it is based on professional ethics, given that Law 41/2002, of 14 November, which is the basic regulation governing patient autonomy, rights and obligations in the field of clinical information and documentation. Article 2 of this law states that “all actions in the medical sphere require, in general, the previous consent of the patients or users”, so that patients must be informed of any variation in their conditions, including the need to inform them of a specific change of treatment. The doctor in charge must decide whether written consent is required, and this will be more necessary to the degree that the result of the intervention is doubtful, as specified by article 10 of the said Law.
Thus in Spain it is not legally possible for a Hospital Pharmacy (HP) to substitute biological drugs, and interchangeability is the responsibility of the prescribing doctor, who has to take ethical and clinical considerations into account, always with the consent of the patient.
Within the context of our maximum collaboration with managers and the HP, doctors have the right to select the medicine we consider to be the best for the patient from among the medicines included in the state financing regime. This capacity is recognised by article 36 of the Constitution, article 4.7 of the Law Regulating the Medical Professions, article 77.1 of Law 29/06 and Decree 2065/1974.
Pharmacovigilance. Prescription using a commercial nameTo ensure traceability, and more specifically the attribution of potential adverse effects, BD must be prescribed using their commercial name. According to article 5.12 of Royal Decree 577–2013 of 26 July, which regulates pharmacovigilance for human use medicines, measures have to be implemented that have the purpose of identifying the name of a medicine and the batch number in notifications that refer to medicine with a biological or biotechnological origin.
Biosimilar controversiesStructural complexity and the manufacturing of biological drugsBiological products, including BSD, are large complex proteins which require transcription machinery and the translation of a live organism, usually a mammal cell, to be produced. BD may have multiple isoforms which differ minimally from each other (microheterogeneity) due to: 1) posttranslational modifications, and 2) minor changes in their manufacturing process, such as different culture media, temperatures or purification techniques.5 This microheterogeneity may influence immunogenicity, and this is associated more closely with clinical efficacy than it is with safety.
To manufacture a BSD the RD has to be exhaustively analysed, using reverse engineering6 to determine its amino acid sequence. BSD developers have to be knowledgeable and have experience in the development and manufacture of BD, and they have to establish their own protocols, given that the manufacturing processes for RD are protected by copyright laws. Even small changes in the manufacturing process may give rise to differences between BSD and RD respecting the level and nature of glycosilation, protein folding or the capacity to interact with other proteins. To gain regulatory approval these modifications must not affect the quality, purity or potency of a BSD, and nor may they cause clinically significant changes in safety or effectiveness in comparison with the RD.7
To create a BSD sophisticated tools and analytical methods are used to prove that there are no structurally relevant differences between the BSD in question and its RD. Nevertheless, a package of comparative analytical data between a BSD and its RD is insufficient for the approval of the BSD if no pre-clinical data (in animals) are supplied, together with clinical data (in humans). All of these data form the “total evidence” used to evaluate the degree of similarity between a BSD and its RD.7–9
To be approved a BSD must pass through a long development route that may be divided into 4 phases:
- 1
Transfect the selected cell line with the DNA that carries the “genetic instructions” to produce the sequence of amino acids of the biological drug.
- 2
Multiple versions of the biological drug are generated in the previous process, each one of which is produced by a “cellular clone”. Scientists evaluate them to select the clone that produces the BSD that is the most similar to the RD.
- 3
The manufacturing process is developed and perfected on an industrial scale to generate a sufficient amount of the BSD for commercialisation.
- 4
After developing the manufacturing process, preclinical and clinical trials take place, The clinical trials are subjected to an accelerated process of clinical evaluation that often involve 2 stages in which the RD is compared with the BSD:
- •
A first stage with healthy volunteers (Phase Ia) or patients (Phase Ib) to confirm that the human body processes the BSD in the same way as it does the RD, i.e., its main objectives are its pharmacokinetics and pharmacodynamics.
- •
A second stage in which a broad set of patients is used to confirm that the BSD has a similar level of efficacy, safety and immunogenicity as the RD.
- •
The extrapolation of the efficacy and safety data of a BSD in one pathology to the other indications of its RD is a key concept in the guidelines of the regulatory agencies, and it has to be scientifically justified.7,10,11 This must be based on the “totality of the evidence”, for which non-clinical data are required, together with physicochemical and functional analysis that justifies them.7,10–12 The process of comparison to establish biosimilarity is undertaken using a step by step approach and through an exhaustive process of comparability, in which any relevant difference found has to be justified. Initially non-clinical studies must be performed, the first step in which are in vitro studies which have to be sensitive enough to detect any difference between the biological activity of the BSD and the reference biological product. The second step consists of evaluating the need to undertake in vivo studies, which have to determine whether, for example, there are effects mediated by monoclonal antibodies that are undetectable by in vitro studies. Nevertheless, these may be unnecessary if the comparability of the in vitro studies was satisfactory. The third step would be to carry out the in vivo studies themselves, where the approach will depend on the need for additional information, and no studies on toxicity in humans will be necessary.3 However, when the RD has different mechanisms of action depending on the diseases for which it is indicated,7,10,11 it may be necessary to perform new functional studies and/or pharmacodynamic studies to gain regulatory approval by extrapolation. These additional data will supply increased surety that the BSD will have similar clinical safety and efficacy for the extrapolated indications as those that were obtained for the indication for which the CT was performed.12
It is not possible to demand greater predictability in terms of safety and efficacy in a BSD than it is in any other new product that reaches the market.13 In fact, the extensive preclinical analysis that is demanded for BSD, including those which are evaluated to grant extrapolation, actually reduce the degree of uncertainty about these compounds in comparison with any other new product.
The regulatory agencies have no single approach regarding the extrapolation of indications. They study these on a case-by-case basis and may reach different decisions. For example, the original infliximab is approved for rheumatoid arthritis (RA), ankylosing spondylitis (AS), psoriasis, psoriatic arthritis (PsA), ulcerative colitis (UC) and Crohn’s disease (CT).14 The infliximab BSD, CT-P13 and SB2, have only been studied using direct comparison trials with the original in patients with AR15,16 and AS.17 The EMA, FDA and Korean regulators approved CT-P13 (Remsima® and Inflectra®) for all of the indications of the RD.18,19 Nevertheless, Japan only approved them for RA, CT and UC,20 and the Canadian agency did not initially support data extrapolation for CT or UC, demanding clinical evaluation for these pathologies.21,22 The Canadian agency recently approved the indications for CT, fistulising CT and UC for this infliximab BSD based on the accumulated evidence for its similarity with the original respecting quality, mechanism of action, disease physiopathology, safety profile, dosage regime and clinical experience with the original.23
As has been remarked, extrapolation is a scientifically logical extension of the BSD concept. Nevertheless, opinions against this have been expressed24–26 that centre on safety, above all at paediatric age27 and clinical efficacy in inflammatory intestinal diseases in which the mechanism of action differs from that of RA.28 The initial decision of the EMA and FDA to accept the extrapolation of infliximab BSD to inflammatory intestinal diseases respecting their efficacy, safety and immunogenicity is now supported by observational studies29–31 and 2 recent systematic reviews32,33 which conclude that there are no differences in terms of efficacy, safety or immunogenicity respecting the RD.
Interchange/substitutionOne of the most controversial questions is whether a BSD can substitute the RD in patients with controlled or stable disease without losing efficacy or increasing adverse effects. Two EU reports for professionals and for patients evaluate this aspect. The report for professionals states: “Decisions on interchangeability and/or substitution depend on the competent authorities in each country and they fall outside the scope of the EMA and the CHMP”.34 The report for patients states: “All decision on therapeutic interchange (switching from one medicine to another) must be taken by your doctor after asking you and taking into account any possible established practice on the use of biological medicines in your country. For any question in connection with a change from biological medicine to another, patients must ask their doctor, pharmacist or specialised nurse”.35
The EMA and the EC have conjointly published a guide for healthcare professionals, in which it states that: “the EMA does not interchangeability, switching or the substitution of a reference drug by its BSD. These are the competence of the Member States of the EU.”4 Thus the authorisation of a BSD does not mean that it is considered to be interchangeable with its RD. Interchangeability (the medical practice of switching from one medicine to another with which it is expected to achieve the same effect, at the initiative of or with the approval of the prescribing doctor) unlike substitution (the practice of substituting the prescribed medicine for another equivalent one by a pharmacist in the act of dispensing, without previous consultation or without the knowledge of the prescribing doctor) must be a clinical decision by the doctor, reached individually and based on scientific evidence and with the knowledge and consent of the patient. However, in Anglo-Saxon medical literature the term interchange (switching) may refer to: 1) Changing one RD for another one, 2) Changing a RD for a BSD, 3) Changing a BSD for another BSD and 4) a Medical switch vs. a Non-Medical switch. This adds more variability and controversy to the interpretation of the results, and from a clinical viewpoint, each one of the meanings of the term switching may have different consequences.
Following the approval by the EMA of the first BSD of a monoclonal antibody in September 2013, multiple extension studies of CT for different BSD have evaluated the switch from a RD to its BSD, although these studies have different designs.36 The majority of them evaluated the transition from a RD to a BSD, while a minority studied the simple change (BSD to RD and RD to BSD after the blind phase) or multiple changes between BSD and RD. According to the authors in question, the majority of the studies are not sensitive enough to exclude possible risks associated with the practice of switching between BD that are very similar to each other (but not identical).
In the extension of CT-P13 after 102 weeks, the randomised patients assigned to Remicade® changed to CT-P13, without any differences being observed in measurements of efficacy, adverse effects or in the formation of antidrug antibodies (ADA).37 Nor were any differences found in the PLANETAS extension in terms of efficacy, safety and immunogenicity, although there was a numerical disparity with a higher percentage of adverse effects and the formation of ADA in the group that switched from the RD to the BSD: 4.8% vs. 3.3% and 27.4% vs. 23.3%, respectively.38 In the extension of the SB2 (Flixabi®), at 78 weeks, at week 54 the patients in the initial Remicade® group were re-randomised to carry on with this drug or switch to the SB2, while the patients randomly assigned to SB2 initially continued taking it. A total of 94 patients switched to SB2 and 101 continued with Remicade®; in week 78 the safety, efficacy and immunogenicity profile was comparable in all 3 groups (SB2/SB2, Remicade®/SB2 and Remicade®/Remicade®).39
In the extension phase of the SB4, which included 254 patients, 126 continued with SB4 and 119 switched from Enbrel® to SB4, and the clinical response, PROS and radiological progression data were comparable.40 In the EGALITY study multiple switching took place between Enbrel® and GP2015. The study was divided into 4 periods: a) screening, b) treatment period 1 (week 0–12) 1:1 randomised assignment to GP2015 (n = 264) or Enbrel® (n = 267), c) treatment period 2 (week 13–30) in which the patients with an improvement of at least 50% in the PASI were randomly assigned to continue with GP2015 or an alternative treatment sequence between GP2015 and Enbrel® every 6 weeks, and d) extension phase (week 31–52) in which the patients continued receiving the treatment administered during the last 6 weeks of period 2. In weeks 30 and 52 no differences were observed in the primary outcome variable, PASI 75 or the other variables. Nor was it found that the multiple switches between Enbrel® and GP2015 affected efficacy, and there were no differences in immunogenicity and safety.41 Finally, a review of the BSD for adalimumab found no differences in terms of efficacy, safety or immunogenicity between Humira® and its BS.42
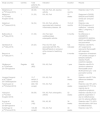
Multiple real-life studies have been published with switches between Remicade® and CTP13 (Table 2)43–54 and between Enbrel® and SB4 (Table 3),55–66 without detecting losses of efficacy, safety problems or changes in the immunogenicity profile; some studies evaluated the rates of BSD persistence, and this was similar to those of the reference cohorts. A loss of efficacy was only detected in one study (23 patients) in which the switch took place due to regulatory reasons;47 the authors concluded that the switch should not take place due to economic or legal reasons. They do not refer to the possible role of the nocebo effect.67,68
Real-life studies with switching from Remicade® to CTP13.
| Study (country) | Centres | Total patients (No. switched) | Indication | Duration | Results |
|---|---|---|---|---|---|
| Nikiphorou et al.43(Finland 2015) | 1 | 39 (39) | RA, AS, PsA, JIA, reactive arthritis | 13 months | Retention rate 71.8% |
| Malaiya et al.45(United Kingdom 2016) | 1 | 31 (30) | RA, AS, PsA | 12 weeks | Response to DAS28, BASDAI and PSARC similar pre- and post-switch) |
| Abdalla et al.44(Ireland 2016) | 1 | 34 (34) | RA, AS, PsA, arthritis associated with intestinal inflammatory disease, JIA | 15.8 ± 6 months | Retention rate 85.2%Suspension (2 ineffective, 1 adverse effect, 1 pregnancy, 1 others) |
| Batticciotto et al.46(Italy 2016) | 3 | 31 (36) | AS, PsA, SpA undifferentiated, enteropathic arthritis | 6 months | No difference in DAS28PCR, ASDASPCR, BASDAI, BASFI and MASES pre- and post-switch |
| Gentileschi et al.47(Italy 2016) | 1 | 23 (23) | PsA, AS, RA, SpA associated with IID, RA, BehcetPatients in remission at the moment of switching | Not indicated | Retention rate 69.6% (5/7 suspensions returned to Remicade® with response) |
| Benucci et al.48(Italy 2017) | 3 | 41 | SpA | 6 months | Suspension of treatment 1/41 (3%)No changes in BASDAI; BASDFI, ASDAS PCDR, DASPCR, MASES |
| Glintborg et al.49(Denmark 2017)Prospective | Register | 802 (802) | RA, AS, PsA | 1 year | Retention rate 83.4% (similar to reference Remicade® cohort 86.8%)Similar pre – and post switch activity scores |
| Vergara-Dangond et al.50(Spain 2017)Retrospective | 1 | 13 (7 switch; 6 continue) | RA, AS, PsA | 24 weeks | Retention rate 85.7%No differences between groups |
| Tweehuysen et al.52(Holland 2018) | 4 | 222 (192 accept) | RA, AS, PsA | 24 weeks | Retention rate 76% (24% suspended CT-P13, the majority due to subjective symptoms) |
| Holroyd et al.54(United Kingdom 2018) | 1 | 59 (59) | RA, AS, PsA, enteropathic arthritis | 52 weeks | Retention rate 86.2% (8 patients suspended: 4 due to ineffectiveness and 4 to adverse effects) |
| Avouac et al.51(France 2018)Prospective | 1 | 260 (162) | RA, AS, IID | 34 weeks | Retention rate 77% (23% discontinuation, 80% due to ineffectiveness) |
| Scherlinger et al.53(France 2018) | 1 | 100 (89 accept) | RA, AS, PsA | 33 weeks | Retention rate 72% |
JIA: juvenile idiopathic arthritis; PsA: psoriatic arthritis; RA: rheumatoid arthritis; AS: ankylosing spondylitis; IID: inflammatory intestinal disease; SpA: spondyloarthritis.
Real-life studies with switch from Enbrel® to SB4.
| Study (country) | Centres | Total (No· switched) | Indication | Duration | Results |
|---|---|---|---|---|---|
| Szlumper et al.55(United Kingdom 2017) | 1 | 109 (103) | RA, AS, PsA, SpA, JIA, Bechet | 6 months | No difference in efficacy /safetyAnnual saving 112,410 £ |
| Rabbits et al.56(United Kingdom 2017) | 1 | 83 (70 switch, 13 naïve) | RA, ES, PsA, JIA, SpA, SpA undifferentiated | NA | Retention rate 84% |
| Sigurdardottir et al.57(Sweden 2017) | 1 | 147 | RA, PsA, AS, JIA, SpA undifferentiated | 8 months | Retention rate 86%No difference in efficacy or safety |
| Holroyd et al.58(United Kingdom 2017) | 1 | 92 | RA, AS, PsA | 6 months | Retention rate 91%No difference in efficacy or safety |
| Hendricks y Hørslev-Petersen61(United Kingdom 2017) | 1 | 85 | RA, AS | 4-8 months | Retention rate 89%No loss of efficacy vs. basal |
| Dyball et al.59(United Kingdom 2017) | 1 | 35 | RA | NA | Retention rate 83%No difference in efficacy /safetyAnnual saving 26,400 £ |
| Glintborg et al.60(Denmark 2017) | Register | 1,5842030(1623) | RA, PsA, SpA | 3 months1 year | Retention rate 83%No differences in activity pre-/post-switch |
| Tweehuysen et al.66(Holland 2018) | 1 | 625 | RA, AS, PsA | 6 months | Retention rate 90%90 % (88–93) |
| Shah et al.62(United Kingdom 2018)Prospective | 1 | 151 | RA | NA | No differences DAS28, HAQ, EQ5D pre and post-switchSaving 500,000 £ |
| Patel et al.63(United Kingdom 2018) | 1 | 193(168) | RA | NA | 11% (n = 18) returned to the RD (7% ineffective, 4% intolerance)Saving 200,000 £ |
| Smith et al.64(United Kingdom 2018) | 1 | 217 | NA | 9 months | Retention rate 95.4%10 returned to RD (4 acute episode, 5 adverse effects, 1 no data) |
| Attipoe et al.65(United Kingdom 2018) | 1 | 355 | RA, AS, PsA | NA | Evaluated predictive response factors (> disease duration, less clinical support and adverse effects) |
| Scherlinger et al.53(France 2018) | 1 | 52(48) | AS, RA | NA | 92% patients accepted86% patients good experience of change5%-15% discontinued treatment |
JIA: juvenile idiopathic arthritis; PsA: psoriatic arthritis; RA: rheumatoid arthritis; AS: ankylosing spondylitis; SpA: spondyloarthritis.
Of the real-life studies in which Remicade® was switched to CTP-13, the study by Glintborg et al.49 included the highest number of patients. In this study Remicade ® was obligatorily replaced by CTP-13. 802 patients were included (403 RA, 279 AS and 120 PsA) without observing changes in the measurements of efficacy evaluated 3 months beforehand, at the moment of switching and 3 months after the switch. The retention rates were somewhat lower in the cohort with CT-P13 (84.4%) versus the historic cohort with Remicade® (86.6%), with a difference in the adjusted absolute risk of 3.4% (P = .03). The authors indicate that the difference is not necessarily attributable to CT-P13, as rather it may represent a “nocebo effect”.67,68 Of the studies that switched from Enbrel® to SB4, the one with the highest number of patients is BIO-SPAN, with a total of 642 patients (433 RA, 128 PsA and 64 AS). This study evaluated survival and efficacy in the switch from Enbrel® to SB4, using patients treated with Enbrel® en 201466 as the comparative historic cohort. The rate of persistence after 6 months of SB4 was 90% (CI 88%-93%) versus the historic Enbrel® cohort of 92% (CI 90%-96%). When they were compared with the historic cohort, the patients who switched to SB4 had a higher relative risk of treatment discontinuation: 1.57 (CI 95% 1.05–2.36). These differences are considered to be clinically irrelevant and 2 explanations for them were suggested: a) in the SB4 cohort there were more suspensions due to more subjective causes, due to the nocebo effect,67–69 and b) the slight differences between the cohorts may be explained by a calendar time distortion, which associate treat-to-target strategies with greater adherence in the year 2016 than they do in 2014.
The study which seems to confirm the efficacy and safety of the switch from the RD to its BSD is NOR-SWITCH,70 a non-inferiority trial that lasted for 52 weeks. Its primary aim was to evaluate whether the switch to CTP-13 was not inferior to Remicade®, in connection with no worsening of the disease, while also evaluating safety, immunogenicity and the efficacy of CT-P13 versus Remicade®. 482 patients were included (155 CT, 93 UC, 91 spondyloarthritis, 77 RA, 30 PsA and 30 psoriasis) in stable treatment with Remicade® during at least 6 months. They were randomly assigned to continue with Remicade® (n = 241) or to switch to CTP-13 (n = 241). Standardised outcome measurements were used to define worsening, or doctor-patient consensus that the disease had worsened and that there was a need for a change of treatment. The no-inferiority margin was 15%. 26% of the patients with Remicade® worsened vs. 30% of the group with CTP-13. The adjusted treatment difference with a 95% CI was −12.7% at 3.9%, which is within the set non-inferiority margin, indicating that CTP-13 is not inferior to Remicade®. There were no differences in adverse effects or efficacy variables. Respecting immunogenicity, no differences were found between the groups in terms of the appearance of ADA at any time of the study, corresponding to 11% of the Remicade® group vs. 13% of the CTP-13 group. This study was not designed to compare treatment strategies individually in each one of the diseases. Nevertheless, the study result supports the idea that Remicade® may be interchangeable with its CTP-13 without compromising efficacy and safety.
A recent study records suspension rates due to adverse effects and the total rate of suspension in double-blind studies of different BSD, and no differences were found in the open phases of these trials in which switching took place from the RD to be BS.69
NOR-SWITCH and DANBIO60,70 study data, together with educational and training activities for doctors and patients are of key importance for the acceptance and use of BSD. Examples exist that show training and information lead to the implementation of interchangeability policies. In Southampton Hospital, digestive system specialists and rheumatologists developed a shared gains program to implement the switch from the RD to CTP-13.71,72 The program was agreed by managers, nurses, doctors and patients. These agreements lead to cost savings and makes it possible to invest in medical services while maintaining clinical results.71 However, in spite of the published evidence there are still doubts about aspects such as whether the data from these studies can be extrapolated to other BSD, or the degree of efficacy and safety when multiple switching occurs between several BSD of the same RD.36,73
ImmunogenicityThe majority of BD are able to elicit an immune response. The immune response is complex and the innate as well as the acquired immune system may contribute to the development of adverse effects. Nevertheless, the formation of ADA is generally referred to as immunogenicity. Although immunogenicity may cause the development of acute and potentially dangerous immunological reactions,74 ADA do not usually clinically affect safety, while their presence is associated with the partial or total loss of drug efficacy.
A range of factors influence immunogenicity. Some are linked to the patient, genetics, age, basal disease and concomitant treatments, while others are linked to the product used, dose, form of administration, manufacturing processes, formulation, stability and impurities.75,76
In the year 2015 the EMA updated the guides for the evaluation of the immunogenicity of therapeutic proteins produced by biotechnology (Table 1). This guide shows the strategies and methodology used in studying the immunogenicity of BD and the requirements for comparison of the immunogenicity of BSD in comparison with RD. Comparative studies are classified as indispensible for the development of BSD, and all trials must measure the immunogenicity of BSD as well as the RD. The incidence and nature of the development of these antibodies must be described, including their crossed reactivity, their target epitopes and whether or not there is neutralising activity. Although BSD are produced in different cells lines form the RD, no significant differences have been found in their immunogenicity in pivotal CT or in the extension studies of the different BSD that have been approved.37,70 However, the regulatory agencies specify that pharmacovigilance is indispensible to detect events that may occur during commercialisation (Table 1).
The frequency of immunogenicity described in the registration trials for BSD and RD is usually higher and differs from the frequency of the same described during their use in clinical practice.77 The reason for this discrepancy is the use of more sensitive techniques and less drug interference during clinical practice. This amounts to an advance from the methodological and scientific points of view, although it does not mean that it has any clinical repercussion. Thus the mere description of the frequency with which ADA appear without describing its pharmacokinetic effect on the therapeutic protein is hardly relevant.
The analyses of the PLANETRA and PLANETAS studies describe differences in the production of ADA between patients with RA and AS. Although these are linked to the different doses and concomitant treatments used in the trials, there were no differences here between CTP-13 and Remicade®.24 Two independent groups have shown that the anti-infliximab antibodies are able to recognise CTP-13 and that they aim above all for the region of the antibody that is associated with binding to the TNF, without being influenced by the glycosylated residues of the IgG moledcule.78,79 Likewise, study of the ADA which appeared in the PLANETRA and PLANETAS studies showed that they have a crossed reaction, meaning that the immunological response to Remicade® and to CTP-13 aims against the same epitopes.80 A recent study published to determine whether anti-infliximab ADA in patients with inflammatory intestinal disease display crossed reaction with Remicade®, CT-P13 and SB2, found that the ADA in patients treated using CTP-13 or patients who had switched from Remicade® to CT-P13 showed a complete crossed reactivity with CT-P13 and SB2. These findings indicate that the immunodominant epitopes of the RD and CT-P13 are equally present in SB2.81
The phase iii trial that compared SB4 with Enbrel® found ADA in at least one determination in 2 patients (0.7%) in the group treated with SB4 and 39 (13.1%) in the Enbrel® group. The ADA appeared very soon in both cases, from week 2 to week 8, and practically all of them disappeared in week 12. The ADA were transitory and not neutralising.82 The presence of ADA had no clinical relevance and did not affect levels of the drug. Similar results have been described with GP2015.83
The opinion of patients respecting the use of biosimilar drugsBSD have the potential to improve patient care by increasing healthcare system efficiency, optimising access to BD and broadening the available treatment options. As a result, the availability of BSD may lead to broader use of BD, and this may give rise to better health outcomes.84
Surveys about BSD and RD have been conducted in patients, groups of patients, doctors and the general population,85–91 to obtain basic information on the use and awareness of BSD. In general the patients believe that RD are superior in terms of efficacy compared with BSD, although no differences in their safety are perceived.85 These studies show that the information supplied to patients by their doctors and associations is usually higher quality, making it possible for them to play a more active role in therapeutic decision-making, including the use of BSD.
In a survey of patients and rheumatologists, 49% of the patients knew what a BSD is, due to information they had received from patient associations, above all. The doctors perceived more differences to exist between RD and BSD than did the patients. It was found that, independently of the price, patients trusted the opinion of their doctor when selecting one or the other, while the rheumatologists, if the price were the same, would prefer to prescribe the original drug. The most important aspects for the patients when switching from an original drug to a BSD were: the opinion of the doctor and that the efficacy of the BSD was proven for their disease. On the other hand, 28% of the rheumatologists surveyed believed that a BSD and an original drug should never be interchangeable, while 39% did not support the extrapolation of indications.92 In another survey, the majority of the specialists were prepared to start with a BSD in a naïve patient for biological drugs, although only a minority considered switching from an original drug to a BS.93
BSD are here and it is foreseeable that they will be increasingly used. We must therefore be able to answer the questions of our patients:
- none-
What is a BSD and what do regulatory agencies require to approve them?
- none-
To what degree are they effective and safe?
- none-
What happens if you switch from a RD to a BSD?
- none-
Why is a BSD cheaper than the RD?
- none-
Could a HP switch from a RD to a BSD without the consent of the doctor and patient?
It is obvious that doctors, scientific societies, patients, patients’ associations, HP and managers will all have to work in achieving this educational goal. It is therefore indispensible that educated and informed patients play an active role in the therapeutic decision-making process. The document for patients that was recently published by the EU should be made available to all of the patients who are going to receive these drugs.35
The position of the Spanish Society of RheumatologyIn the SER we express our complete commitment to the sustainability of the healthcare system of our country, and we support the measures that, without reducing the quality of care, have the aim of ensuring its continuity. The EMA authorisation to commercialise the BSD of original biological drugs will give rise to an excellent opportunity to progress in the efficiency of medical care, and it will improve rheumatic patient access to biological therapies.
Within this new scenario of an increasing offer of biological drug therapies, in the SER we consider it to be indispensible to preserve the freedom of prescription of doctors to prescribe drugs according the individual characteristics and circumstances of each patient, without forgetting the economic aspects that derive from this action.
A BSD is a BD that contains a version of the active substance of an original biological product that has already been authorised, and to be approved it have to show that its variability and any difference respecting the RD has no effect on its safety or efficacy. Thus once a BSD has been authorised, the regulatory agencies guarantee that there are no significant differences in it compared to the RD in terms of quality, efficacy and safety.
The SER therefore wishes to make it clear about BSD drugs, that:
- 1
A BSD drug is a BD that has proven biosimilarity in in vitro studies with its reference drug, so that it is indistinguishable from it in terms of quality, biological activity, safety and efficacy, within the framework of direct comparison by randomised double-blind clinical trials.
- 2
The selection of the target to block and the active substance is the responsibility of the prescribing doctor, and this selection must be decided exclusively within the context of the doctor-patient relationship, taking the characteristics of the disease to be treated into consideration together with any relevant comorbidities and after informing the patient.
- 3
Once the target to be blocked has been selected together with the active substance, the choice of an innovative drug or a BSD is the responsibility of the doctor, and this should be decided exclusively within the context of the doctor-patient relationship. This decision will take safety and cost-effectiveness into account.
- 4
Switching from a biological drug to its BSD must take place exclusively at the decision of the prescribing doctor, with the consent of the patient. In the case of patients with stable disease a switch from the RD to its BSD may be acceptable, although this decision must be individualised and it is subject to the consent of the patient.
- 5
There is currently no scientific evidence on the efficacy and safety of switching between the different BSD of a single reference drug. This should be taken into account when information patients of this, if the prescribing doctor advises switching between BSD.
- 6
The SER understands that hospital institutions have to guarantee that all of the BD and BSD financed by the medical authorities in our country to treat rheumatic diseases are available in all National Health System hospitals.
- 7
BSD are subject to the same safety monitoring as their RD, so that it is necessary to favour their inclusion in the specific pharmacovigilance registers which are currently being developed. The SER has broad experience in these registers and offers to undertake these safety studies.
- 8
The traceability of the BD is an aspect of quality that makes it possible to assign suspicions of adverse reactions to specific batches and products. BSD are currently assigned the same international common denominator as the original drug, so that prescription has to take place using the commercial name, to achieve suitable traceability.
- 9
If the RD has more than one indication, the extrapolation of indications must be justified according to EMA standards.
- 10
The optimum use of BSD requires continuous dialogue and interaction between doctors, HP, patient associations and regulatory bodies, with the aim of preserving patients’ right to health and with the aim of offering them high quality, effective and safe products.
- 11
This position of the SER will be regularly updated in the light of new evidence, and it is estimated that the next update will occur within 2 years.
M. Á. Abad Hernández. He has worked with and received fees from MSD, Pfizer, Celgene, Kern, Novartis, Biogen, Sandoz, Amgen and Janssen, as a consultant, speaker, researcher and/or member of advisory boards.
J. L. Andreu. Fees for speaking from Abbvie, Antares, GSK, MSD, Nordic, Novartis, Sanofi and UCB. Fees for research projects and consultancies: Abbvie, Amgen, AstraZeneca, Biogen, Cellgene, Celltrion, Fresenius Kabi, Gebro, Pfizer and Regeneron.
A. Balsa Criado. He has received fees for presentations, advisory work or research funds from Abbvie, MSD, Pfizer, UCB, Roche, BMS, Nordic, Sandoz, Lilly, Sanofi and Novartis.
F. Díaz-González. For speaking: Pfizer, MSD, Lilly, Janssen, BMS and Roche. For scientific consultancy: Lilly, Novartis, Pfizer, Amgen, Biogen and Celgene. For research projects: MSD, Abbvie, Roche and Novartis.
J. V. Moreno Muelas. Gebro, Janssen, MSD, Pfizer and Sanofi.
R. Queiro Silva. He has worked with and received fees from Abbvie, MSD, Pfizer, Celgene, UCB, Lilly, Novartis and Janssen as a consultant, speaker, researcher and /or member of advisory boards.
J. J. Gómez-Reino. Advisory boards and consultancies: Abbvie, Biogen, BMS, Gebro, GSK, Lilly, Novartis, Pfizer, Roche, R-Pharma, Sandoz, Sanofi and Regeneron; Conferences: Abbvie, BMS, Celgene, Janssen and Janssen, Lilly, MSD, Pfizer, Roche, Sanofi and UCB; grants: MSD, Pfizer, Roche and UCB.
Please cite this article as: Abad Hernández MÁ, Andreu JL, Balsa Criado A, Díaz-González F, Moreno Muelas JV, Queiro Silva R, et al. Actualización del Documento de posicionamiento de la Sociedad Española de Reumatología sobre fármacos biosimilares. Reumatol Clin. 2021;17:160–169.


