








VEXAS (Vacuoles, E1 enzyme, X-linked, Autoinflammatory, Somatic) syndrome is an adult-onset autoinflammatory syndrome characterized by somatic mutations in the UBA1 gene and is considered the prototype of hematoinflammatory diseases. Patients with VEXAS syndrome exhibit inflammatory and hematological manifestations that can lead to clinical diagnoses such as relapsing polychondritis, polyarteritis nodosa, Sweet syndrome, and myelodysplastic syndrome. Diagnosis requires bone marrow evaluation to identify cytoplasmic vacuoles in myeloid and erythroid precursors. However, genetic confirmation of mutations in UBA1 is necessary. Treatment is challenging and often involves glucocorticoids and immunosuppressants with variable responses. Hypomethylating agents and allogenic haemopoietic stem cell transplant are considered promising therapies. Prognosis is influenced by genetic and clinical factors. The aim of this review is to provide an overview of the pathogenesis, clinical presentation, treatment, and prognosis of VEXAS syndrome for the Latin American medical community.
El síndrome de VEXAS (Vacuolas, enzima E1, ligado al X, Autoinflamatorio, Somático) es un síndrome autoinflamatorio de inicio en la edad adulta, caracterizada por mutaciones somáticas en el gen UBA1, y se considera el prototipo de enfermedad hematoinflamatoria. Los pacientes con síndrome de VEXAS exhiben manifestaciones inflamatorias y hematológicas que pueden conducir a diagnósticos clínicos como policondritis recidivante, poliarteritis nodosa, síndrome de Sweet y síndrome mielodisplásico. El diagnóstico requiere la evaluación de la médula ósea en búsqueda de vacuolas citoplásmicas en precursores mieloides y eritroides. Sin embargo, la confirmación genética de las mutaciones en UBA1 es necesaria. El tratamiento es un desafío y a menudo incluye glucocorticoides e inmunosupresores con respuestas variables. Las terapias hipometilantes y el trasplante alogénico de células progenitoras hematopoyéticas se consideran terapias prometedoras. El pronóstico es influido por factores genéticos y clínicos. El objetivo de esta revisión es proporcionar una visión general sobre la patogénesis, presentación clínica, tratamiento y pronóstico del síndrome de VEXAS para la comunidad médica Latinoamericana.
VEXAS syndrome (acronym for Vacuoles, E1 enzyme, X-linked, Autoinflammatory, Somatic) is an autoinflammatory syndrome identified in the late 2020s by Beck et al.1 Using a genotype-focused screening methodology, 25 male patients with adult-onset inflammatory disease and cytopenia were found to carry missense genetic variants in the UBA1 gene, which codes for the ubiquitination activating enzyme E1, a primary regulator of the ubiquitin-proteosome system. Mutations in the chromosome X-linked UBA1 gene were identified as monogenic, somatic, and restricted to cells of the myeloid and erythroid lineage.1 Patients with VEXAS syndrome have a combination of inflammatory and haematological manifestations. The most common manifestations include skin lesions, fever, chondritis, arthritis, pulmonary infiltrates, macrocytic anaemia, thrombocytopenia, and venous thrombosis. Due to the complexity and diversity of the clinical manifestations, its management and treatment requires the collaboration of several medical specialists.
VEXAS syndrome is considered the prototypical haemato-inflammatory disease, a term used to describe those diseases caused by somatic mutations in blood cells that present with systemic inflammation, disturbances in the bone marrow, and are premalignant conditions that may lead to myeloproliferation, myelodysplasia, or lymphoproliferation.2,3
The aim of this review is to provide an overview of the pathogenesis, clinical presentation, treatment, and prognosis of VEXAS syndrome.
Pathogenesis and geneticsThe UBA1 gene is located on chromosome Xp11.23 and encodes for the ubiquitin E1 activating enzyme considered the main initiator of activation, conjugation, and binding of ubiquitin to protein substrates for degradation by the ubiquitin-proteosome system.4,5UBA1 is expressed in a nuclear isoform (UBA1a) of 1,058 amino acids and a cytoplasmic isoform (UBA1b) of 1,018 amino acids which, unlike the nuclear isoform, initiates translation at p.Met41.6 Both isoforms consist of five domains (Fig. 1). The N-terminal region consists of the inactive adenylation domain (IAD), which flanks the first catalytic cysteine domain (FCCD). Towards the C-terminal end is the active adenylation domain (AAD), in which the second cysteine catalytic domain (SCCD) is located. The ubiquitin fold domain (UFD) that allows UBA1 to conjugate E1 comprises the C-terminal end of the protein.7

The pathogenesis of VEXAS syndrome has been linked to loss of UBA1 expression as a consequence of somatic loss-of-function mutations in the IAD and AAD domains.1,8 Mutations in the IAD domain are considered pathogenic and are associated with global loss of ubiquitination, unfolded protein response, increased cellular stress, and activation of innate immune pathways.1,9–11 A high proportion of mutations occur in exon 3 within the IAD domain. The most frequent are found at codon 41, where translation of UBA1b initiates, leading to methionine substitution: p.Met41Thr (c.122 T > C), p.Met41Val (c.121A > G), and p.Met41Leu (c.121A > C). Mutation in p.Met41 causes translation initiation to shift to p.Met67 and, consequently, a decrease in the cytoplasmic UBA1b form and an increase in the dysfunctional UBA1c enzyme as a product of the mutation.1 As a result, innate immune pathways are abnormally activated, leading to hyperproduction of inflammatory cytokines, including tumour necrosis factor α, interleukin (IL)-1, IL-6, IL-8, interferon-inducible protein (IFN)-γ and IFN-γ. These cytokines have been shown to be elevated in the serum of patients, even during clinically inactive periods, and in animal models. This immune disturbance is responsible for the various clinical manifestations present in the syndrome.1 Furthermore, although the mutations occur at the same genomic position, the type of amino acid substitution affects the cytoplasmic translation of UBA1 in a heterogeneous manner. The presence of UBA1c in the cytoplasm leads to increased loss of UBA1b. For example, carriers of the p.Met41Val variant show two-fold lower cytoplasmic levels of UBA1b compared to those with the p.Met41Leu/Thr variants, because there is more UBA1c. These variations at the molecular level correlate with clinical outcomes, as the p.Met41Val genotype has been found to be associated with decreased survival.12 In the same domain, mutations c.118-1G > C; c.119-1 G > C, c.118-2A > C, which occur at the splice site of exon 3 and whose effect is the same as that of mutations in p.Met41,13,14 have been identified. Poulter et al.14 identified a second mutation in the IAD domain. The p.Ser56Phe mutation (c.167C > T) does not have an effect on the decreased expression of UBA1b or the formation of UBA1c; however, it has an effect on the catalytic activity of UBA1, causing reduced ubiquitination. In vitro studies show that the mutation produces a thermolabile UBA1b enzyme with temperature-dependent decrease in its catalytic activity. It is hypothesised that the p.Ser56Phe variant could lead to preferential inactivation of the cytoplasmic UBA1b isoform in cells or alternatively cells may be more sensitive to reductions in UBA1b activity.
Other variants have recently been described in exons 14 and 16 located in the AAD domain: p.Glu441Ter, p.Gly477Ala, p.Ala478Ser, p.Asp506Gly, p.Asp506Asn, p.Asp585Ala, p.Asp585Glu, and p.Ser621Cys, which have been categorised as variants of uncertain significance (VUS). These variants have been shown not to block UBA1 synthesis but to reduce the catalytic function of the cytoplasmic and nuclear isoforms by 40%–90%. The p.Asp506Gly and p.Asp506Asn variants affect UBA1 function by interfering with ATP binding and all specifically alter ubiquitin transfer from UBA1 to the E2 enzyme.8,15,16 Mild autoinflammatory manifestations are another notable finding in VUS carriers. Moreover, carriers of p.Ala478Ser, p.Asp585Ala, and p.Asp585Glu have been reported to have a similar proportion of vacuolated precursors as patients with VEXAS syndrome.16
The UBA1 mutations responsible for VEXAS syndrome were initially described as somatic or postzygotic, limited to cells of the myeloid and erythroid lineage.1 As a result, UBA1 variants can be detected in sites where these cells are present, such as peripheral blood and bone marrow, or in affected tissues where there is infiltration of these cells, such as skin or muscle.1,17,18 In contrast, UBA1 variants have not been observed in the lymphoid lineage or in fibroblasts of affected patients.1 A recent Spanish study identified UBA1 mutations in the nails of patients with VEXAS syndrome. This indicates that UBA1 variants are not limited to myeloid and erythroid precursors. Since nails are part of the ectoderm, it is possible that the mutation occurred at an early stage of embryonic development.19 This new evidence has conceptual implications for the pathogenesis of the syndrome, suggesting that the disease could manifest at any age. So far, however, it has only been diagnosed in adult patients, highlighting the need for further studies to investigate this aspect.
EpidemiologyBecause it has been recently characterised, the incidence and prevalence of VEXAS syndrome have not been fully defined. Only one study has reported the prevalence of the disease.20 This study used DNA from 163,096 individuals participating in the Geisinger MyCode Community Health Initiative, a biobank in Pennsylvania developed to study precision medicine. In this study, pathogenic UBA1 variants were found in 11 individuals (9 males and 2 females), all of whom had clinical manifestations consistent with VEXAS syndrome (100% penetrance). Although 5 of the 11 individuals did not meet the criteria for a diagnosis of definite rheumatological or haematological disease, all had macrocytic anaemia and/or thrombocytopenia. The prevalence of VEXAS syndrome-causing mutations was 1 in 13,591 unrelated individuals, 1 in 4,269 men over 50 years, and 1 in 26,238 women over 50 years.20 These results suggest that VEXAS syndrome may have a higher prevalence than some systemic vasculitides, such as granulomatosis with polyangiitis (1/18,000) and polyarteritis nodosa (1/33,000), and a similar prevalence to myelodysplastic syndromes (MDS) (1/14,000).20
Regarding the age group affected, the seminal work by Beck et al.1 described the average age at onset of symptoms as 64 years, although onset can vary between 40 and 85 years.21 A case of VEXAS syndrome that started at 23 years has recently been identified, suggesting the possibility of early onset of the disease, as proposed by the findings of the Spanish cohort.22 Although all 25 characterised patients were male, fewer than a dozen cases of VEXAS syndrome have been documented in women.23–28 Most of these cases had congenital or acquired monosomy X. An interesting observation from the published studies is that no cases have been reported in black patients.
Clinical manifestationsThe clinical manifestations of VEXAS syndrome are diverse and include both inflammatory and haematological manifestations (Fig. 2). It is common for patients to meet the criteria and to have been diagnosed with other rheumatological, dermatological, and/or haematological diseases (Table 1), prior to a definitive diagnosis of VEXAS syndrome. For example, in the US National Institutes of Health (NIH)/University of Leeds cohort, the most frequent clinical diagnoses were relapsing polychondritis in 52% of cases, myelodysplastic syndrome (MDS) in 31%, undifferentiated febrile syndrome in 23%, and Sweet's syndrome in 22%.12

Clinical diagnoses of patients with VEXAS syndrome.
| Rheumatological |
| Relapsing polychondritis1 |
| Polyarteritis nodosa1 |
| Giant cell arteritis1,29 |
| ANCA associated vasculitis30–32 |
| IgA vasculitis33 |
| Cryoglobulinaemic vasculitis31 |
| Behçet’s syndrome34 |
| Polymyalgia rheumatica20 |
| Rheumatoid arthritis35 |
| Spondyloarthritis36 |
| Dermatomyositis20 |
| Systemic lupus erythematosus37 |
| Undifferentiated autoinflammatory/fever syndrome12,25,29 |
| IgG4-related disease38,39 |
| Sarcoidosis20 |
| Dermatological |
| Sweet syndrome1 |
| Cutaneous polyarteritis nodosa40 |
| Eosinophilic pustular folliculitis40 |
| Erythema nodosum41,42 |
| Lupus tumidus42 |
| Haematological |
| Myelodysplastic syndrome1,43 |
| Multiple myeloma1,43 |
| Monoclonal gammopathy of undetermined significance43 |
| Monoclonal B-cell lymphocytosis43 |
| Chronic myeloid leukaemia24,44 |
| Myelofibrosis20 |
Recurrent fever is one of the most common features of VEXAS syndrome, occurring in a range of 65%–100% of cases.1,24,45,46 Although its pattern is not specific, it usually manifests during periods of disease activity and with increased levels of acute phase reactants. Treatment with corticosteroids usually has a good response, and if not, other possible causes, such as concurrent infection, should be sought.21,47 It is often accompanied by other symptoms, such as nocturnal diaphoresis, malaise, and fatigue. In addition, weight loss has been observed in a percentage of cases ranging from 31% to 56%.1,24,45,46 It has also been observed that up to 34.5% of patients with VEXAS syndrome may present lymphadenopathies in different locations, more frequently at the mediastinal level.46,48
ENTNasal and/or auricular chondritis affects a high percentage of patients, ranging from 46% to 100% (Fig. 3A).1,24,46 Although there are similarities between idiopathic relapsing polychondritis (PR-I) and VEXAS-associated relapsing polychondritis (PR-V), some notable differences have been identified. For example, a French study involving 95 patients (40 with PR-I and 55 with PR-V) showed that patients with PR-I tend to be younger (44 vs. 66 years) and have a lower prevalence of fever (10% vs. 60%), while nasal cartilage is more frequently affected (70% vs. 47%) compared to patients with PR-V.49 Another study by Ferrada et al.45 compared the clinical characteristics of 85 patients with PR-I and 13 with PR-V, observing a higher prevalence of airway chondritis (44% vs. 0%) and costochondritis (85% vs. 0%) in patients with PR-I compared with patients with PR-V. In addition, sensorineural hearing loss has been reported in up to 29% of cases.12 Other less frequent manifestations are submandibular sialadenitis and oral aphthosis.39,50

Manifestations of VEXAS syndrome. (A) Auricular chondritis. (B) Erythematous and oedematous plaques and papules suggestive of Sweet’s syndrome. (C) Septal panniculitis with significant necrosis, fibrosis, and scant perivascular lymphocytic inflammation. (D) Neutrophilic and granulomatous vasculitis affecting medium-sized arteries. F) Simple CT scan of the orbits in coronal section showing evidence of previous orbital myositis, with central hypodensity of the superior rectus muscles (arrows). (G) Chest CT showing bilateral pulmonary infiltrates. (H) Characteristic vacuoles in myeloid precursor cells in bone marrow aspirate.
Skin manifestations have been reported in up to 84% of cases of VEXAS syndrome, which can be divided into non-vascular and vascular lesions.12,24
The most common lesions are firm, painful papules and nodules, with an erythematous-violaceous and oedematous appearance, usually appearing on the extremities and trunk, and less frequently on the face, and may clinically suggest erythema nodosum.40,41,51 In 24% of cases these lesions may be pruritic.40 A local reaction at the anakinra injection site, which may be severe, is commonly reported.1,24,46 In addition, other less frequent manifestations such as livedo reticularis, livedo racemosa, vesicles, bullae, urticariform lesions, and ulcerations have been reported.21,40
A study in France found that neutrophilic dermatosis was the most common histopathological finding in these lesions, and many of these patients had a diagnosis of Sweet syndrome before being diagnosed with VEXAS syndrome (Fig. 3B).1,17,40 Other histopathological patterns observed are septal panniculitis (Fig. 3C), or leukocytoclasia without other findings of vasculitis.40,41
VasculitisThe presence of vasculitis has been described in 20%–64% of cases of VEXAS syndrome and is considered a secondary vasculitis of vessels of variable calibre, as it can affect small, medium, and large vessels.13,21,24,52 In fact, in the initial NIH cohort, some patients had a previous clinical diagnosis of systemic vasculitis, especially polyarteritis nodosa and giant cell arteritis.1 Patients with VEXAS may present with atypical symptoms and patterns of vasculitic involvement that do not fully fit the classification of known primary systemic vasculitides.21
Leukocytoclastic vasculitis is the most common form of vasculitis, seen in 26%–38% of patients, although other types of cutaneous small-vessel vasculitis such as lymphocytic, eosinophilic, urticarial, and bullosa vasculitis have also been described.1,24,29 As mentioned above, VEXAS syndrome can present with cutaneous vasculitis of medium-sized vessels simulating polyarteritis nodosa (Fig. 3D).1,52
Although vasculitis similar to ANCA associated vasculitis has been observed, its presentation is not classic and it does not respond satisfactorily to conventional treatment with rituximab or cyclophosphamide.26–28 In addition, arterial/periarterial thickening has been observed mainly at the carotid level, as well as in the thoracic and iliac aorta.23,24,53 Other forms of vasculitis that have been reported are IgA vasculitis, cryoglobulinaemic vasculitis, and presentations similar to Behçet's syndrome.31,33,34
OphthalmologicalOphthalmological manifestations have been observed in 24%–46% of cases and include ocular inflammatory and orbital manifestations.12,13,24,31 Ocular inflammatory manifestations include episcleritis, scleritis, uveitis, conjunctivitis, chemosis, choroidal detachment, and retinal vasculitis.21,22,24,31,39,49,54 However, orbital manifestations include periorbital and orbital oedema, which have been described in a range of 9%–30% of cases.12,24 Orbital structures that have been affected on imaging studies include the eyelids, lacrimal glands (dacryoadenitis), preseptal and orbital fat (cellulitis), extraocular muscles (orbital myositis), and optic nerve sheath (perineuritis) (Fig. 3F).21,39,46,54–58 Angioedema has also been described in the skin of the face, involving the eyelids.39 It is important to note that orbital swelling is often mistaken for infectious cellulitis.21,54
JointArthralgias have been reported in up to 28% and arthritis in up to 58% of patients diagnosed with VEXAS syndrome.12,24 It usually manifests as an oligoarticular or polyarticular condition affecting small and large joints in both the upper and lower extremities.21 Although there is as yet no information assessing the presence of long-term radiographic changes, it appears that the arthritis is not erosive. Only one case of erosive arthritis and one case of radiographically confirmed sacroiliitis have been reported in a patient with HLA-B27 positivity.35,36
PulmonaryPulmonary manifestations of VEXAS syndrome can be highly variable. Some patients may present with mild symptoms, such as cough and dyspnoea, while others may require mechanical ventilation.45,48 Tomographic findings are common (between 72% and 100% of cases) and the main patterns described include ground glass opacities (present in 87% of cases), consolidation (49%), reticulation (38%), nodules (47%), mediastinal adenopathy (58%), and pleural effusion (53%) (Fig. 3G).48 Some tomographic patterns described include non-specific interstitial pneumonia, bronchiolitis obliterans, and organised pneumonia.21 In cases where lung biopsy has been performed, lymphocytic or neutrophilic alveolitis, neutrophilic vasculitis, or organised pneumonia have been reported.1,46,59 It is important to emphasise that lung involvement can occur within a short period after the start of prednisone tapering.
Other inflammatory manifestationsLess frequently, VEXAS syndrome can also affect other organs. Peripheral nervous system involvement has been reported in 14.7% of cases, gastrointestinal tract involvement in 14%, splenomegaly in 13.8%, orchitis in 12%, kidney in 9.5% (mainly interstitial nephritis), hepatomegaly in 7.8%, pericarditis in 4.3%, and myocarditis in 2.6%.12,24,46 In addition, aseptic meningitis, myofasciitis, and necrotising myositis have been reported.1,58
Haematological manifestationsObiorah et al.43 described the main haematological manifestations of VEXAS syndrome in 18 patients, which include macrocytic anaemia (100%), thrombocytopenia (50%), lymphopenia (80%), monocytopenia (50%), and to a lesser extent neutropenia (13%). In larger cohorts, macrocytic anaemia has been reported in up to 97% of cases and thrombocytopenia in 83% of cases.12 In the peripheral blood smear, mild to moderate macrocytosis, young forms of the granulocytic series, vacuolated, hypogranular, hyposegmented neutrophils, and some with pseudo-Pelger-Huet-like morphology were observed.43,60
Bone marrow aspirate and bone biopsyIn the first series of VEXAS syndrome, the presence of vacuoles in the cytoplasm of the granulocytic and erythroid series was described as typical (Fig. 3H). However, this is not pathognomonic for the disease, and other causes such as zinc intoxication, copper deficiency, alcoholism, and MDS must be ruled out.42,43,60,61 On the other hand, Obiorah et al.43 described that the proportion of myeloid and erythroid precursors with vacuoles was 15%, with a median of 5–7 vacuoles per cell, characterised by being round, small, and well-defined. These vacuoles are mainly located in myeloid or erythroid precursors. To a lesser extent, they can be found in monocytes, eosinophils, plasma cells, and megakaryocytes. The absence of vacuoles in lymphocytes is explained by the fact that the presence of the mutation prevents lymphocytes from surviving. Beck et al.1 detected the mutation in haematopoietic progenitor, megakaryocytic-erythroid progenitor, myeloid progenitor, and lymphoid progenitor cells. In addition, in rare cases, patients with VEXAS syndrome may present without vacuolisation.62
Three histopathological patterns in bone marrow have been described in patients with VEXAS syndrome:
- 1
Morphological findings in patients presenting with unexplained cytopenia/clonal cytopenia of uncertain significance. Hypercellularity (87.5%), increased megakaryocytes (50%), myeloid hyperplasia, and a myeloid to erythroid ratio that increases with time, and dysplasia in any of the three cell lines but in less than 10% of the cells.43,60
- 2
Myelodysplastic syndrome. Patients with VEXAS syndrome may also develop MDS, which has been reported in different series from 24% to 63% using the WHO 2016 criteria.1,12,23,45 These cases are characterised as low-risk according to the IPSS-R, with less than 5% blasts and 50% with a normal karyotype.8 Next-generation sequencing in 9 cases from the original Beck series showed that only 6 had recurrent somatic mutations, including MLL-PTD, DNMT3A, CSF1R, SF3B1, and EZH2, and only one case had two co-existing mutations.1 The presence of the UBA1 mutation suggests that it could be a "driver mutation" in MDS and should be included in the myeloid mutation panel.
- 3
Plasma cell neoplasia and lymphoproliferative diseases. Multiple myeloma, monoclonal gammopathy of uncertain significance, monoclonal lymphocytosis B, and chronic lymphocytic leukaemia have been reported.24,43
Haemophagocytic lymphohistiocytosis has also been reported as a complication of VEXAS syndrome.63
ThrombosisVenous thrombosis may occur in 36%–56% of cases. Arterial thrombosis occurs in less than 10% of cases. Thromboses usually occur early in the first two years after the onset of inflammatory symptoms.43,64 Although the exact cause of thrombosis is unknown, the inflammatory process plays an important role. Transcriptome profiling of blood cells from patients with VEXAS syndrome reveals the expression of inflammatory genes.1 The levels of von Willebrand factor, antithrombin III, protein C and S in patients with VEXAS syndrome are usually found to be normal.43 High levels of factor VIII were found in five patients, of whom only two had thrombosis. A correlation between factor VIII levels and C-reactive protein has been reported. The thromboelastogram was normal in two cases.43,64 In addition, 44% positivity for lupus anticoagulant has been reported, in some cases persisting after 12 weeks. However, the presence of anti-cardiolipin and anti-β2-glycoprotein I antibodies is uncommon.43 Currently, the role of lupus anticoagulant in VEXAS syndrome is uncertain, and it is not known whether it should be considered a true antiphospholipid syndrome.
DiagnosisIn terms of laboratory tests, most patients will have elevated C-reactive protein (CRP), erythrocyte sedimentation rate (ESR), and ferritin. These markers may vary depending on the degree of disease activity.1,24 Positive antinuclear antibodies can be found in about 16% of cases, and positive rheumatoid factor in 20% of cases.1,48 Other antibodies are generally absent in VEXAS syndrome; however, cases have been reported in which patients present with a clinical diagnosis of ANCA associated vasculitis and positivity for anti-myeloperoxidase or anti-proteinase 3 antibodies in the context of VEXAS syndrome.31,32
When VEXAS syndrome is suspected, it is important to look for cytoplasmic vacuoles in myeloid and erythroid precursors in the bone marrow aspirate, if available, as they are present in most cases, as mentioned above.2
Finally, although clinical features, laboratory parameters and bone marrow biopsy are important to identify those with suspected VEXAS syndrome, the diagnosis requires genetic confirmation.1 UBA1 mutations have been described in DNA obtained from peripheral blood mononuclear cells, bone marrow biopsies, or skin biopsies from neutrophilic dermatoses and muscle.1,17,18 Currently, UBA1 mutation testing is mainly restricted to research laboratories in some academic institutions, but is now more available commercially in the United States and in some countries in Europe. When ordering the test, it is important to ensure that the sample is not being tested for a germline mutation in UBA1, which is associated with X-linked infantile spinal muscular atrophy and will not confirm the presence of the somatic mutation.21 It is important to know the test methodology used in UBA1. Most patients with VEXAS have allele frequencies of UBA1 variants>20%, and therefore Sanger sequencing should be sufficient to detect most cases.1 In addition, because mutations in UBA1 are not limited to codon 41, sequencing of the entire UBA1 gene should be requested.65
Screening of patients with suspected VEXAS syndromeVEXAS syndrome should be considered a possible diagnosis in individuals presenting with inflammatory symptoms unresponsive to conventional treatments or dependent on glucocorticoids, and who also have progressive haematological anomalies, especially macrocytic anaemia, and thrombocytopenia (Fig. 4). This syndrome is more common in men over 40 years of age, and therefore the age and gender of the patient should be considered when suspecting this disease.2,21 Specifically, the populations most likely to have a positive UBA1 mutation are those with clinical diagnoses most frequently reported in patients with VEXAS syndrome, such as relapsing polychondritis, Sweet’s syndrome, or MDS.
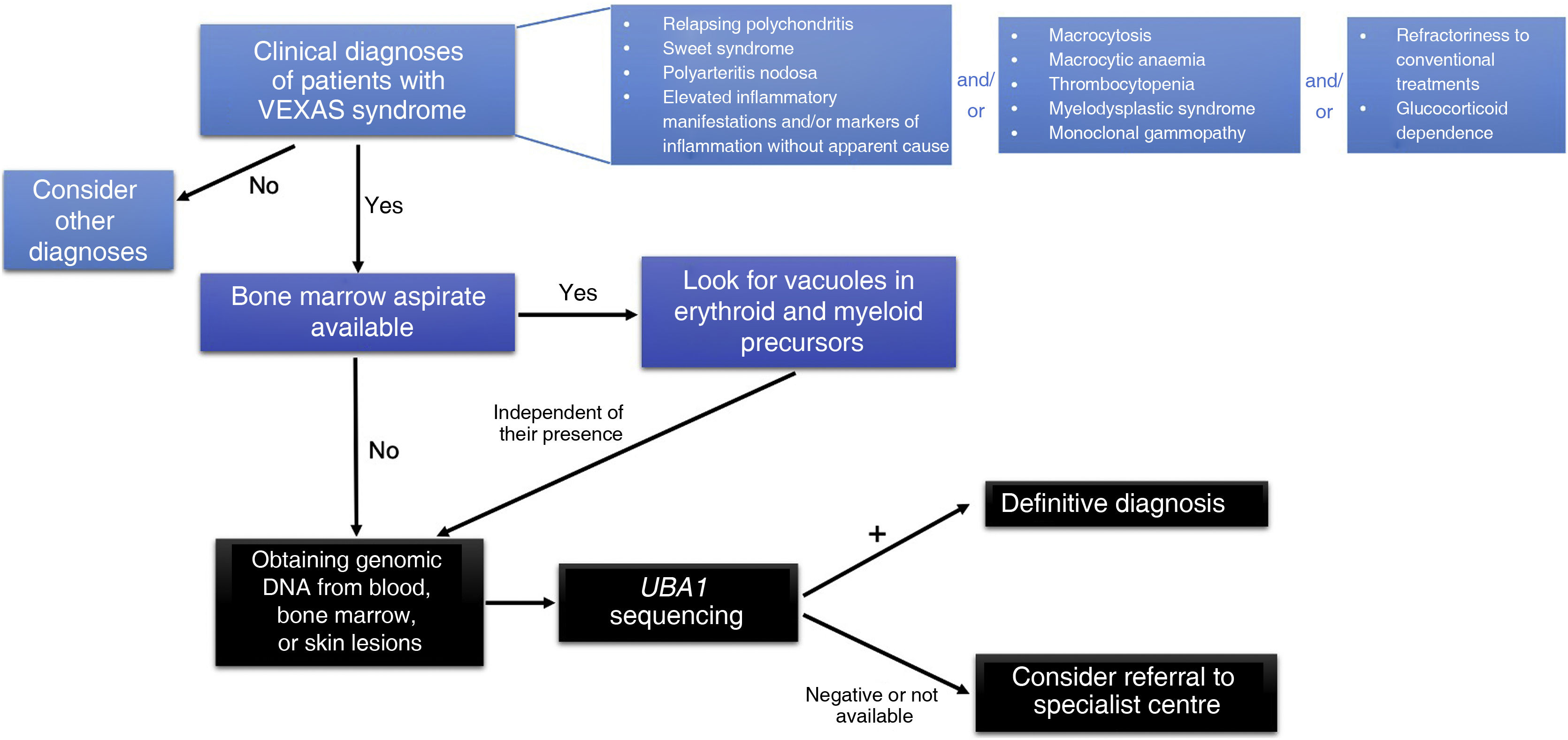
Ferrada et al.45 have developed an algorithm to identify possible cases of VEXAS syndrome in patients with relapsing polychondritis. In a male with a clinical diagnosis of relapsing polychondritis and a mean corpuscular volume (MCV) greater than 100 fL and/or less than 200,000 platelets/μl, genetic analysis for mutations in UBA1 should be considered. This algorithm has demonstrated a sensitivity of 100% and a specificity of 96% in identifying patients with PR-V.
In addition, in men presenting with neutrophilic dermatoses suggestive of Sweet’s syndrome, especially if associated with macrocytic anaemia, the likelihood of VEXAS syndrome is high.17,44
Another study found a high positivity rate for UBA1 in patients with systemic vasculitis of any calibre using the following criteria: (1) recurrent fever and at least one of the following: cutaneous involvement, pulmonary infiltrates, ear or nasal chondritis, or venous thromboembolism; (2) CRP levels >20 mg/l and at least one of the following: macrocytic anaemia (haemoglobin value <12 g/dl and MCV > 100 fl), thrombocytopenia (<130,000/μl), neutropenia (<1.500/mm3), or a haematological diagnosis of MDS, and (3) failure to respond to at least one synthetic or biologic disease-modifying antirheumatic drug and continued need for glucocorticoid therapy.32
In patients with MDS who present with inflammatory manifestations or have been diagnosed with an autoimmune disease, a diagnosis of VEXAS syndrome should be considered and mutation testing for UBA1 is recommended. Two studies have investigated this issue and found that, among the patients tested, a prevalence of UBA1 mutation of 12% and 33% was detected.25,29
Finally, the presence of vacuoles in myeloid and erythroid precursors in patients with MDS or inflammatory diseases is another scenario that should raise the suspicion of VEXAS syndrome.42,61 One study has shown that identification of ≥10% neutrophil precursors in a bone marrow aspirate with >1 vacuole has a sensitivity and specificity of 100% for detecting patients with VEXAS syndrome.66
TreatmentTreatment of VEXAS syndrome has two main goals: eradicating cells with the UBA1 mutation and inhibiting the inflammatory pathways and cytokines involved.67 This is a challenge due to multi-organ involvement, heterogeneity in presentation, frequent association with haematological malignancies, dependence on glucocorticoids, and refractoriness to conventional immunosuppressive therapies.68,69
There are currently no standardised treatment algorithms, and therefore recommendations are based on a limited number of retrospective studies and case series.8 Therefore, the design of clinical trials, the definition of meaningful (beyond "time to next dose or next treatment") and reliable response and efficacy criteria, and the identification of predictive biomarkers are needed.
Glucocorticoids and conventional immunosuppressantsGlucocorticoids at doses of 20−40 mg/day of prednisone are the most effective anti-inflammatory therapy in patients with VEXAS syndrome, with transient and dose-dependent effect (difficulty in reducing the prednisone dose below 10−20 mg/day).21 Long-term use is associated with infectious and cardiovascular adverse events, and therefore other glucocorticoid-sparing drugs have been tried. The choice of therapy depends on the predominance of inflammatory or haematological manifestations.8 Calcineurin inhibitors (cyclosporine and tacrolimus) in combination with glucocorticoids have demonstrated clinical and biological response in inflammatory symptoms associated with VEXAS syndrome in a small group of patients.47,69
Other immunosuppressants, including methotrexate, cyclophosphamide, azathioprine, and mycophenolate mofetil, have been used in the treatment of these patients, with little evidence of their efficacy as monotherapy.47,52,54
Biological drugsTherapy with anti-IL-1 drugs (anakinra and canakinumab) has shown mixed results, mostly associated with discontinuation due to site reactions or recurrence of symptoms.8 Anti-TNF alpha agents (infliximab, etanercept, golimumab, and adalimumab) have been associated with ineffectiveness or partial response.47,70 Treatment with anti-IL-6 (tocilizumab) has shown transient efficacy in some cases (median time to next treatment of 8 months), with improvement in skin and haematological manifestations, transfusion requirements, and glucocorticoid dose.55,71 In other patients, no response has been observed, and there are two case reports of intestinal perforation in patients with VEXAS syndrome who received tocilizumab.8,21,47,55 Other drugs that have been used in patients with VEXAS syndrome include anti-CD20 (rituximab), anti-IL-17 (secukinumab), and anti-IL-12/IL-23 (ustekinumab) therapy, with no evidence of efficacy.47 In the case of rituximab use, patients with VEXAS syndrome may experience prolonged CD19 recovery time, which may increase the risk of opportunistic infections.
Small moleculesTreatment with Janus kinase (JAK) inhibitors, including ruxolitinib, baricitinib, upadacitinib, and tofacitinib, has been associated with clinical response after the first month of therapy (especially ruxolitinib), including glucocorticoid dose reduction, transfusion independence, skin improvement, and normalisation of CRP levels. However, side effects such as serious bacterial and viral infections, haematological toxicity, and thrombotic events have been reported.8,21,69,72
Other anti-inflammatory and immunomodulatory drugsThere are reports of the use of hydroxychloroquine, dapsone, colchicine, lenalidomide, bortezomib, abatacept, and intravenous immunoglobulin in patients with VEXAS syndrome, without clear clinical benefit.8,43,69
Hypomethylating agentsAzacitidine and decitabine are DNA methyltransferase inhibitors and are used in the treatment of patients with VEXAS syndrome who develop MDS.68,69 Treatment with azacitidine, a drug approved for the treatment of high-risk MDS, has demonstrated improvement in inflammatory symptoms, reduction in glucocorticoid and transfusion requirements, haematological response, normalisation of bone marrow disturbances, and near complete eradication of mutated clones, with variations in response according to the presence of MDS-related mutations.8,47,73,74 These agents are a therapeutic option for patients with high-risk VEXAS syndrome associated with MDS who are not candidates for allogeneic transplantation.69,74
Allogenic haematopoietic stem cell transplantation (HSCT)This strategy is based on the potential eradication of cells with the UBA1 mutation. However, it is associated with high morbidity and mortality due to the risk of developing graft-versus-host disease and infectious complications, therefore it is important to consider factors such as age, functional status, and the presence of comorbidities.69 Fewer than 20 cases of HSCT in VEXAS syndrome have been reported, most with complete response, but with very short follow-up times.75
In the absence of other curative therapies, HSCT could be considered early in glucocorticoid-dependent patients with active and refractory inflammation, concurrent MDS, and good functional, mental and nutritional status.69,75–77 This intervention may also be considered in patients with progressive cytopenias or other high-risk data, such as the presence of the p.Met41Val mutation, transfusion dependence, requirement of >20 mg/day of prednisone for symptom control, failure of two or more lines of therapy, or presence of other clonal mutations in the marrow (e.g. DNMT3A, TET2).8 A phase II study to evaluate the efficacy of HSCT in patients with VEXAS syndrome is currently ongoing (ClinicalTrials.gov Identifier: NCT05027945).
Supportive careSupportive care in patients with VEXAS syndrome plays an extremely important role; it must be individualised and consider the risk of MDS, bleeding, and thrombosis. It includes blood transfusions, growth factors, erythropoietin-stimulating agents, thrombopoietin mimetics (eltrombopag), and prophylaxis against opportunistic infections.8,69
Anticoagulation is considered in patients with VEXAS syndrome who develop thrombotic events, duration depends on the presence of other risk factors. Thromboprophylaxis should also be considered in hospitalised patients, with prolonged immobilisation or recent surgery, in the absence of contraindications. There is no strong evidence on the type of anticoagulant, vitamin K antagonists being preferred in those with VEXAS syndrome and antiphospholipid antibody positivity, or in the presence of arterial thrombosis.64
Because patients with VEXAS syndrome are generally older and have long-term steroid exposure, it is crucial to prevent, assess, and treat associated conditions. In this regard, measures for the diagnosis, prevention, and treatment of osteoporosis are recommended in these patients. Other empirical measures, such as prophylaxis for Pneumocystis jirovecii and herpes zoster reactivation, should also be considered.
PrognosisBecause it has been so recently characterised, the prognosis of VEXAS syndrome has not yet been fully established. However, it is considered to be a progressive disease that may present with multiple relapses and accumulation of damage from both the disease itself and the treatments used.
Disease-related complications and toxicity from immunosuppressive drugs and glucocorticoids are common causes of death.12,24 Another Dutch cohort reported a 50% mortality at a median follow-up of 4 years, while the French cohort reported a lower mortality rate of 15.5% at a follow-up of 3 years.24,46 A more recent update of the NIH cohort of 83 patients reported a mortality of 25%, with a median survival of 10 years from onset of symptoms to death. In addition, factors associated with mortality, such as having the p.Met41Val mutation or transfusion dependence, were identified in this cohort, while having atrial chondritis was associated with lower mortality.12
In contrast, in a French cohort of patients with VEXAS syndrome, the p.Met41Leu variant was reported to be associated with better survival. Further factors associated with mortality were also identified, such as the presence of gastrointestinal involvement, pulmonary infiltrates, and mediastinal adenopathy.24
Conclusions and perspectivesVEXAS syndrome is a disease that occurs as a result of somatic mutations in myeloid cells, the prototype of a new group of diseases called haemato-inflammatory syndromes. Although initially thought to be a rare disease, its reported prevalence suggests that it is more common than previously thought. This syndrome should be considered in adults, especially those over the age of 40, who present with inflammatory manifestations that do not respond to treatment and/or progressive cytopenias.
The optimal treatment for VEXAS syndrome is currently unknown, but glucocorticoids remain the most useful drugs, although their use is associated with an accumulation of adverse effects. An alternative treatment option to glucocorticoids has not yet been established, although better results have been reported with tocilizumab, azacitidine, and ruxolitinib. In severe cases, HSCT may be a curative option. To date, most cases of VEXAS syndrome have been described in populations in the United States, Europe, Japan, and Australia. In Latin America, only three cases of VEXAS syndrome have been reported.22,54,78 This lack of information about the disease in the region could be due to a lower diagnostic capacity and lack of knowledge about the disease in the medical community. Because VEXAS syndrome is a rare but potentially serious disease, it is important to raise awareness of the disease worldwide, including in Latin America. This could enable earlier detection, accurate diagnosis, and the appropriate management of affected patients.
Conflict of interestsThe authors have no conflict of interests to declare.
The authors would like to thank Silvia Méndez-Flores, Daniel Montante-Montes de Oca, and Jesús Delgado-de la Mora for their valuable support in clinical and histopathological imaging.


