


Given the paucity of data in Latin America and especially in Argentina regarding the epidemiology of SSc, the prevalence of ILD, its course, and particularly the response to treatment, our objective was to evaluate a cohort of SSc patients evaluated in a single University Hospital in Buenos Aires.
Patients/MethodsWe included 152 patients with SSc, followed from disease onset to last pulmonary function test and with at least two PFT and up to 30 months between each.
ResultsSixty-one percent had diffuse SSc (DSSc) and 32% limited SSc (LSSc). The only significant clinical differences between these groups were a higher initial mRodnan score and prevalence of ILD in the DSSc. These also had significantly more anti Scl-70 (Topoisomerase 1) antibodies compared to the LSSC group who had significantly more anti centromere antibodies. The DSSc group also had significantly more extensive damage on HRCT with no differences in terms of imaging patterns.
Comparing patients with and without ILD by HRCT, those with ILD had significantly more extensive damage, significantly more anti Scl-70 antibodies, and significantly fewer anti centromere antibodies than those without ILD.
Patients whose ILD progressed had a smoking history (OR 4.97) and prior immunosuppressive treatment (OR 15.6) (multivariate analysis). Overall disease duration was significantly shorter in those who progressed.
ConclusionsOur SSc population had similar characteristics to those described elsewhere as well as prevalence of ILD and its progression. We found a shorter disease duration, smoking, and prior immunosuppressive treatment to be associated with ILD progression.
La escasez de datos en Latinoamérica, y especialmente en Argentina, sobre la epidemiología de la esclerosis sistémica (SSc), la prevalencia de enfermedad pulmonar intersticial (EPID) y su progresión, llevó a evaluar una cohorte de pacientes con SSc atendidos en un hospital universitario de Buenos Aires, Argentina.
Pacientes/MétodosIncluimos 152 pacientes con SSc, seguidos desde el inicio de la enfermedad hasta el último examen funcional respiratorio (EFR) y con por lo menos dos EFR separados por un mínimo de 30 meses.
ResultadosEl 61% tenían enfermedad difusa (DSSc) y el 32%, limitada (LSSc). Aquellos con DSSc tuvieron significativamente un mayor índice modificado de Rodnan y prevalencia de EPID. Estos también tuvieron significativamente más anticuerpos anti-Scl-70 (topoisomerasa 1) comparados con LSSc, quienes tuvieron significativamente más anticuerpos anti-centrómero. Aquellos con DSSc mostraron significativamente más daño en la tomografía computada de alta resolución (TACAR), pero sin diferencias respecto a patrón de imágenes.
Aquellos con EPID por TACAR tuvieron significativamente más daño, más anticuerpos anti Scl-70 y menos anticuerpos anti-centrómero que aquellos sin EPID.
La progresión de EPID (análisis multivariado) se relacionó con consumo de tabaco (OR: 4,97) y uso previo de inmunosupresores (OR: 15,6). La duración de la enfermedad fue menor en los que progresaron.
ConclusionesNuestra población de SSc tuvo características similares a lo descripto en el resto del mundo, así como la prevalencia y la progresión de EPID. Encontramos una menor duración de enfermedad, el consumo de tabaco y el uso previo de inmunosupresores asociados a la progresión de EPID.
Systemic sclerosis (SSc; scleroderma) is a relatively rare disease whose pathogenesis is characterized by 3 hallmarks: small vessel vasculopathy, production of autoantibodies, and fibroblast dysfunction leading to increased deposition of extracellular matrix. Despite recent advances in certain aspects of disease and further knowledge in some areas of pathophysiology it remains a disabling condition that shortens life expectancy.1
SSc has several clinical variants but essentially presents within two large groups regarding disease-extension: limited cutaneous SSc (lcSSc) where the affected skin is restricted to the distal limbs and face and diffuse cutaneous SSc (dcSSc) where the skin thickening extends proximal to the elbows and may involve the trunk. These two forms may have different degrees of internal organ involvement and be associated with different autoantibodies with different course and prognosis. Therefore, it is important to identify the clinical subset as early as possible.
With the advent of angiontensin converting enzyme inhibitors three decades ago, the principal cause of death which was renal involvement (scleroderma renal crisis) is now controlled more satisfactorily and the principal causes of death today are pulmonary and cardiac.
Pulmonary disease in SSc mainly comprises interstitial lung disease (ILD)-essentially associated with the diffuse form- and pulmonary arterial hypertension (PAH)-usually associated with the limited form of SSc. Over the past 40 years the SSc mortality rate has seen a shift from renal disease to lung involvement (including both ILD and pulmonary hypertension). These have become the primary cause of SSc-related deaths (33% and 28% respectively), replacing SSc renal crisis (with a drop of frequency from 42% to 6% since the awareness of the role of high-dose systemic corticoids in this disease, among other things), whereas the proportion of deaths due to heart disease has not changed significantly over time.2
Cumulative survival of SSc patients from diagnosis is 84.1% at 5 years and 74.9% at 10 years, but when we look particularly into the group of SSc-ILD patients the 10 year survival rate is significantly lower (29–69%) which clearly shows it is important to recognize patients with ILD early and treat them appropriately.3
There are defined risk factors for the development of ILD in patients with SSc: some of them are disease-related like the presence of dcSSc, anti-Scl-70/anti-topoisomerase I antibody and/or absence of anti-centromere antibody 4 and shorter disease duration.6 Others are demographic-related as African–American ethnicity and older age at disease onset. However, none of these risk factors is absolute.
The risk of developing ILD is greatest early in the course of SSc, so much so that particularly in diffuse SSc, the initial approach was to perform a HRCT scan and PFT at diagnosis and annually the first five years. Even though there is some rational knowledge regarding which patient presents an elevated pretest risk of developing SSc-ILD, there is still poor information as to which of these will then progress.5,6
Guler et al. showed different disease behaviour patterns among patients with SSc-ILD, renewing the challenge of finding risk factors and predictors of pulmonary function decline.7 A recent review analyzed the different measures in pulmonary function tests (PFT) used as outcomes for SSc-ILD. It showed that the widespread use of FVC for the Scleroderma Lung Study I and II trials somehow led to a reduction of efforts to identify other possible predictors of SSc-ILD progression, despite the fact that FVC may only weakly reflect the extent of the disease. Ultimately, the best surrogate marker for SSc-ILD onset and progression may be a composite outcome consisting of a combination of two or more measures.8
Over the past decade, several important aspects became apparent regarding ILD as a whole and particularly in SSc, the connective tissue disease more frequently affected and more frequently participating in all drug trials involved when studying ILD in these diseases. The clarification that IPF was not amenable to immunosuppression, the recognition of an ILD in patients without clear-cut connective tissue diseases (IPAF) perhaps responding to immunosuppression, and more recently the demonstration of progression of fibrosis in spite of prior treatment, immunosuppressive or not, have changed our current scenario.9–11 Particularly, the suggestion that a combination of immunosuppression and anti-fibrotic drugs may be useful in some patients has recently been raised.12
Therefore, especially regarding SSc, the early diagnosis of ILD and the identification of possible predictors of progression even under treatment, are very important issues today.
In Latin America and particularly in Argentina, to our knowledge, there is scarce data regarding the epidemiology of SSc, the prevalence of ILD and its course and even less regarding response to treatment.13 A large series from Brazil with almost one thousand patients showed that mortality was greater in males with diffuse SSc and lung and heart disease were the main causes of death.14 A recent report from a University Hospital in Buenos Aires with a cohort of over 200 patients showed an overall prevalence of ILD of 65.3% in the diffuse form and 26.7% in the limited variant. However, no evaluation of progression of ILD was made.15 A study from the centre originating the current data analyzed the response to treatment of 24 patients with SSc and ILD. Eighteen of those had extensive disease as per Goh criteria 16 and 6 had limited lung disease. Twelve of the 18 with extensive disease were treated with cyclophosphamide for a median of 9 months. Although these patients did not improve their lung function, this was stabilized.17
Because of the paucity of overall data in this part of the world, it was our intention to analyze some of the above mentioned aspects in a cohort of SSc patients followed prospectively in a single centre from the Buenos Aires University Hospital (Hospital de Clínicas José de San Martin) evaluated by both rheumatologists and lung specialists.
Our main goal was to describe our SSc population, determine the prevalence of ILD, and see if we could identify differences between patients who progressed and those who did not.
Materials and methodsStudy designWe conducted a retrospective observational cohort study analyzing adult patients with diagnosis of SSc, evaluated by both the Rheumatology and Respiratory Care Divisions.
Patient cohortAdult patients attending both the Division of Rheumatology and Respiratory Care at the University Hospital in Buenos Aires between 2007 and 2018 who fulfilled the American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) classification criteria for SSc were included in the study.18 They were classified as diffuse cutaneous (dc) or limited cutaneous (lc) according to LeRoy's criteria (skin involvement proximal or distal to elbows or knees respectively).19 All patients with at least one HRCT at first visit with us or within 12 months prior to and at least two PFT's within the 30 months we established as follow-up were then included.
Patients with missing data regarding the lung (HRCT, PFTs) or lost to follow-up were excluded from the analysis.
Definition of clinical involvementInvolvement of skinThe maximum extension of skin involvement at any one-time during course of the disease was considered to define subsets (limited vs. diffuse). mRSS score was determined at basal visit in most patients.20 Digital ulcers: active digital ulcers or pitting scars confirmed by a physician at any time during disease course, calcinosis either clinical or radiological, telangiectasias confirmed by a physician at any time during disease course and Raynaud's phenomenon by colour changes usually caused by cold exposure.21
Lung involvementInterstitial lung disease (ILD)Defined as pulmonary interstitial disease observed in high resolution computerized tomography (HRCT) which was performed at initial visit or within 12 months prior.HRCT scans were obtained with 16-slice Aquilion Lightning Toshiba. Patients were examined at end inspiration. The images were evaluated by a general radiologist initially but later re-evaluated by a thoracic radiologist blinded to the clinical data. We took both tomographic diagnoses into account but if there was disagreement, the thoracic radiologist's opinion prevailed.
The functional lung assessment (PFT) was as followsInitial evaluation performed within six months after initial visit included: forced vital capacity (FVC), forced expiratory volume in one second (FEV1), FEV1/FVC, diffusing capacity for the lungs measured using carbon monoxide (DLCO), total lung capacity (TLC) and residual volume (RV). All were performed according to the American Thoracic Society/European Respiratory Society guidelines22 using a Medical GRAPHICS™ device. The results were expressed as absolute and percentages of predicted values. The lower limits of normal considered for this study were <80% of predicted for FVC, TLC and for DLCO, and <0.7 for FEV1/FVC. DLCO value was obtained by the single-breath method and corrected for haemoglobin.
Follow up PFT were considered for evaluation of progression performed up to 30 months after the initial PFT.
Progressors Vs Non ProgressorsWe defined progressors as those showing a decrease in FVC in millilitres of 5% or more and/or a decrease of 10% or more of DLCO (in ml/min/mmHg) values at 30 months. In this case, progressors were defined as per PFT deterioration irrespective of the fact that at initial visit they may not have had ILD as defined by HRCT.
Pulmonary hypertension (PH)Echocardiogram with estimated pulmonary systolic artery pressure greater than 40mmHg or right heart catheterization with mean pulmonary artery pressure at rest over 25mmHg.
Detection of autoantibodiesANA antibodies were detected by immunofluorescence (IMF) using HEp2 cells, particularly for anti centromere antibodies, and a titre of 1/80 or higher was considered positive. Antibodies to Ro (SSA), La (SSB), U1RNP, Sm, and anti Scl-70 (anti-Topoisomerase 1) were detected by double immunodiffusion or ELISA.
Immunosuppressive treatmentTreatment of ILD was decided by each treating physician, according to the extent of the disease, symptoms, and risk of progression so we were not able to obtain precise data as far as these indications.
We considered treatments having been received when there was at least an indication of having received corticosteroids, azathioprine, cyclophosphamide, mofetil mycophenolate or a combination of these.
We then analyzed those who received at least one drug versus those who did not.
Ethical considerationsThe protocol was approved by the Comité de Ética Hospital de Clínicas “Jose de San Martin”, University of Buenos Aires.
Statistical analysisContinuous variables were described with median and quartiles 1 (Q1) and 3 (Q3). Categorical variables were described with frequency and percentage. For analysis of variables regarding SSc population, Chi-square or Fisher's tests were used for categorical variables and Kruskal–Wallis test for continuous variables. To compare variables according to presence or absence of ILD and for ILD progression, Chi-square or Fisher's tests were also used for categorical variables and Wilcoxon for continuous. Assumption of normality of continuous variables was analyzed with the Shapiro–Wilk test.
To determine which variables were associated with ILD progression, a multivariate logistic regression model was adjusted, and the variables were selected stepwise; for significant variables, Odds Ratios (OR) were estimated by comparing the groups and their respective 95% confidence intervals.
For all tests the level of significance was established at 0.05. Analysis was performed using R software.
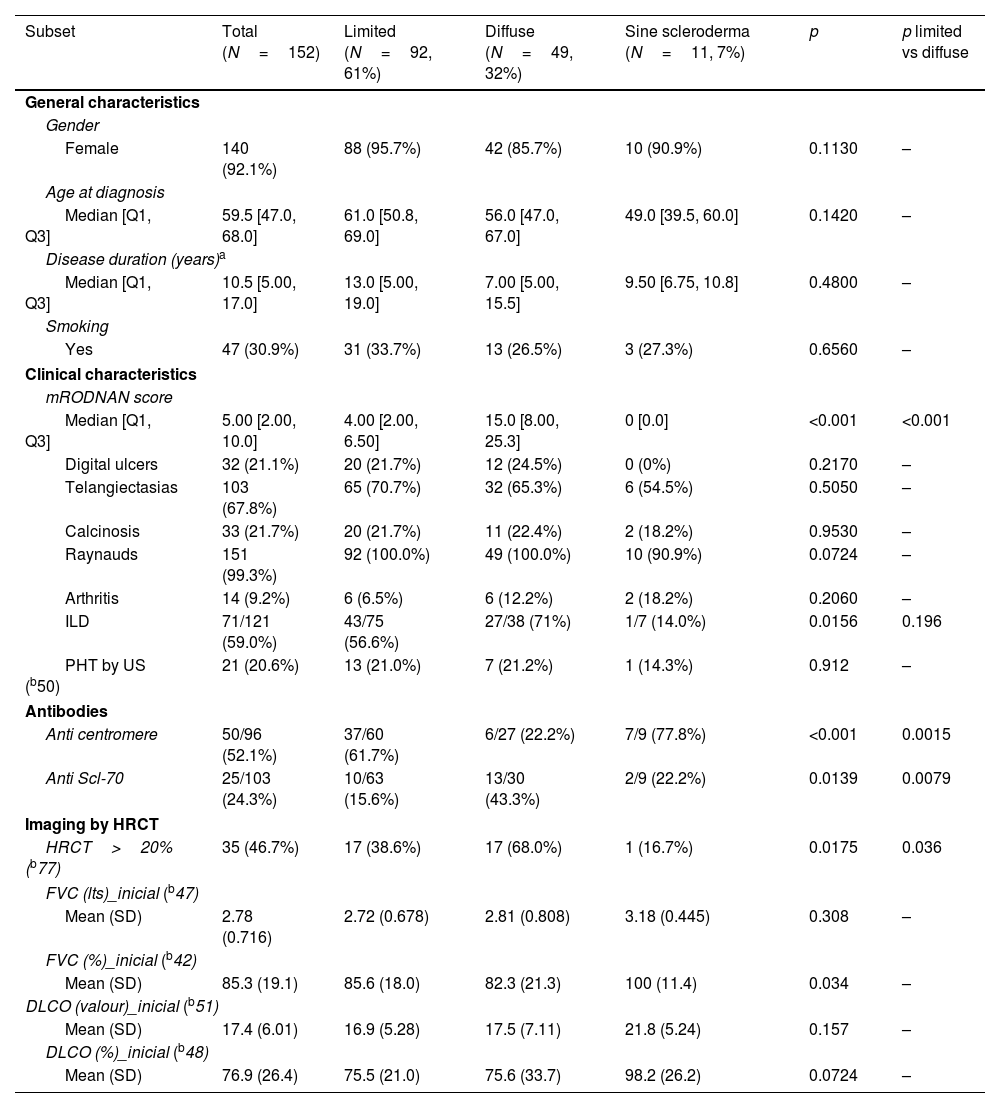
ResultsFig. 1 shows the flow of patients evaluated during the study period. Two hundred and seven fulfilled inclusion criteria. Fifty five were then excluded so as not to confuse results because they had features of overlap with a variety of other connective tissue diseases. One hundred and fifty two then became our study population and their characteristics are shown in Table 1.

General characteristics of our SSc population.
| Subset | Total (N=152) | Limited (N=92, 61%) | Diffuse (N=49, 32%) | Sine scleroderma (N=11, 7%) | p | p limited vs diffuse |
|---|---|---|---|---|---|---|
| General characteristics | ||||||
| Gender | ||||||
| Female | 140 (92.1%) | 88 (95.7%) | 42 (85.7%) | 10 (90.9%) | 0.1130 | – |
| Age at diagnosis | ||||||
| Median [Q1, Q3] | 59.5 [47.0, 68.0] | 61.0 [50.8, 69.0] | 56.0 [47.0, 67.0] | 49.0 [39.5, 60.0] | 0.1420 | – |
| Disease duration (years)a | ||||||
| Median [Q1, Q3] | 10.5 [5.00, 17.0] | 13.0 [5.00, 19.0] | 7.00 [5.00, 15.5] | 9.50 [6.75, 10.8] | 0.4800 | – |
| Smoking | ||||||
| Yes | 47 (30.9%) | 31 (33.7%) | 13 (26.5%) | 3 (27.3%) | 0.6560 | – |
| Clinical characteristics | ||||||
| mRODNAN score | ||||||
| Median [Q1, Q3] | 5.00 [2.00, 10.0] | 4.00 [2.00, 6.50] | 15.0 [8.00, 25.3] | 0 [0.0] | <0.001 | <0.001 |
| Digital ulcers | 32 (21.1%) | 20 (21.7%) | 12 (24.5%) | 0 (0%) | 0.2170 | – |
| Telangiectasias | 103 (67.8%) | 65 (70.7%) | 32 (65.3%) | 6 (54.5%) | 0.5050 | – |
| Calcinosis | 33 (21.7%) | 20 (21.7%) | 11 (22.4%) | 2 (18.2%) | 0.9530 | – |
| Raynauds | 151 (99.3%) | 92 (100.0%) | 49 (100.0%) | 10 (90.9%) | 0.0724 | – |
| Arthritis | 14 (9.2%) | 6 (6.5%) | 6 (12.2%) | 2 (18.2%) | 0.2060 | – |
| ILD | 71/121 (59.0%) | 43/75 (56.6%) | 27/38 (71%) | 1/7 (14.0%) | 0.0156 | 0.196 |
| PHT by US (b50) | 21 (20.6%) | 13 (21.0%) | 7 (21.2%) | 1 (14.3%) | 0.912 | – |
| Antibodies | ||||||
| Anti centromere | 50/96 (52.1%) | 37/60 (61.7%) | 6/27 (22.2%) | 7/9 (77.8%) | <0.001 | 0.0015 |
| Anti Scl-70 | 25/103 (24.3%) | 10/63 (15.6%) | 13/30 (43.3%) | 2/9 (22.2%) | 0.0139 | 0.0079 |
| Imaging by HRCT | ||||||
| HRCT>20% (b77) | 35 (46.7%) | 17 (38.6%) | 17 (68.0%) | 1 (16.7%) | 0.0175 | 0.036 |
| FVC (lts)_inicial (b47) | ||||||
| Mean (SD) | 2.78 (0.716) | 2.72 (0.678) | 2.81 (0.808) | 3.18 (0.445) | 0.308 | – |
| FVC (%)_inicial (b42) | ||||||
| Mean (SD) | 85.3 (19.1) | 85.6 (18.0) | 82.3 (21.3) | 100 (11.4) | 0.034 | – |
| DLCO (valour)_inicial (b51) | ||||||
| Mean (SD) | 17.4 (6.01) | 16.9 (5.28) | 17.5 (7.11) | 21.8 (5.24) | 0.157 | – |
| DLCO (%)_inicial (b48) | ||||||
| Mean (SD) | 76.9 (26.4) | 75.5 (21.0) | 75.6 (33.7) | 98.2 (26.2) | 0.0724 | – |
Sixty one percent of patients had limited SSc and 32% diffuse, with only 12% with sine scleroderma. There were no significant differences in gender, age at diagnosis, disease duration or smoking history between groups in these patients.
Regarding clinical characteristics, aside from a significantly higher mRSS in the diffuse patients (median 15 vs 4 years), the presence of ILD was more frequent (71% vs 56.6%) but did not reach statistical significance. Other characteristics were similar.
With respect to autoantibodies, as was to be expected, anti-centromere antibodies were significantly more prevalent in limited SSc as anti Scl-70 antibodies were in the diffuse variant.
When considering imaging, diffuse patients had significantly more damage extension in HRCT (68%) than the other groups. We did not have sufficient data regarding HRCT patterns to analyze this information.
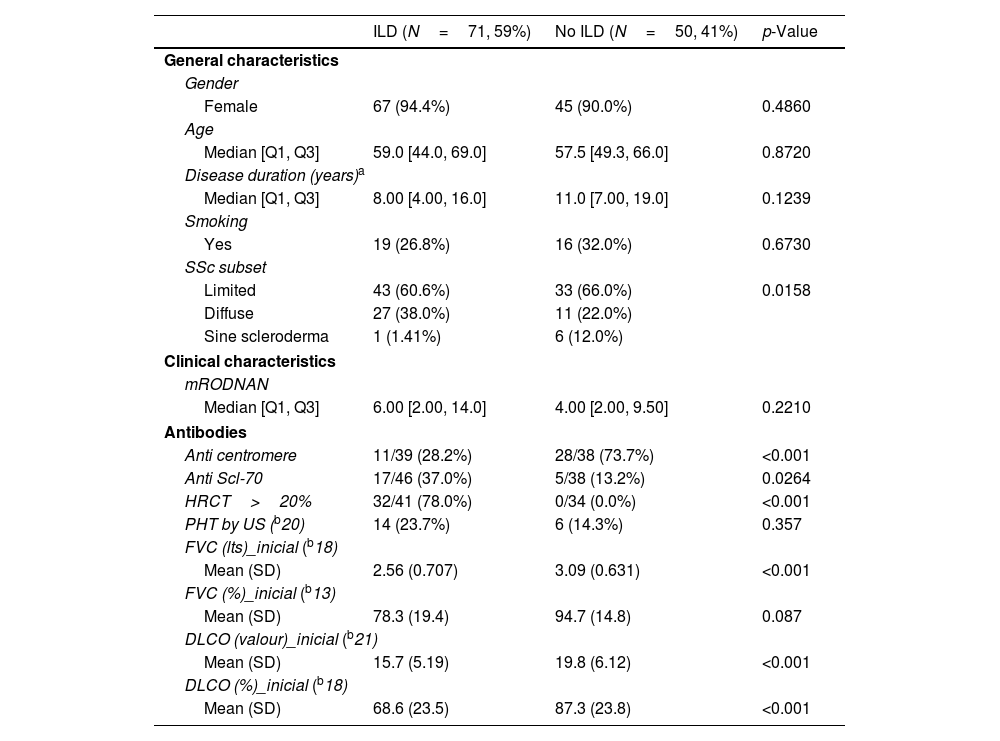
Table 2 shows a comparison between patients with and without ILD as per HRCT definition. There were no significant differences in general characteristics such as gender, age, disease duration or smoking history. As expected, ILD was more prevalent in the diffuse variant and patients with ILD had significantly more extensive damage in HRCT.
Characteristics according to the presence or absence of ILD.
| ILD (N=71, 59%) | No ILD (N=50, 41%) | p-Value | |
|---|---|---|---|
| General characteristics | |||
| Gender | |||
| Female | 67 (94.4%) | 45 (90.0%) | 0.4860 |
| Age | |||
| Median [Q1, Q3] | 59.0 [44.0, 69.0] | 57.5 [49.3, 66.0] | 0.8720 |
| Disease duration (years)a | |||
| Median [Q1, Q3] | 8.00 [4.00, 16.0] | 11.0 [7.00, 19.0] | 0.1239 |
| Smoking | |||
| Yes | 19 (26.8%) | 16 (32.0%) | 0.6730 |
| SSc subset | |||
| Limited | 43 (60.6%) | 33 (66.0%) | 0.0158 |
| Diffuse | 27 (38.0%) | 11 (22.0%) | |
| Sine scleroderma | 1 (1.41%) | 6 (12.0%) | |
| Clinical characteristics | |||
| mRODNAN | |||
| Median [Q1, Q3] | 6.00 [2.00, 14.0] | 4.00 [2.00, 9.50] | 0.2210 |
| Antibodies | |||
| Anti centromere | 11/39 (28.2%) | 28/38 (73.7%) | <0.001 |
| Anti Scl-70 | 17/46 (37.0%) | 5/38 (13.2%) | 0.0264 |
| HRCT>20% | 32/41 (78.0%) | 0/34 (0.0%) | <0.001 |
| PHT by US (b20) | 14 (23.7%) | 6 (14.3%) | 0.357 |
| FVC (lts)_inicial (b18) | |||
| Mean (SD) | 2.56 (0.707) | 3.09 (0.631) | <0.001 |
| FVC (%)_inicial (b13) | |||
| Mean (SD) | 78.3 (19.4) | 94.7 (14.8) | 0.087 |
| DLCO (valour)_inicial (b21) | |||
| Mean (SD) | 15.7 (5.19) | 19.8 (6.12) | <0.001 |
| DLCO (%)_inicial (b18) | |||
| Mean (SD) | 68.6 (23.5) | 87.3 (23.8) | <0.001 |
Patients with ILD had a higher mRSS score but this did not reach statistical significance.
Anti Scl-70 was significantly more prevalent in patients with ILD as opposed to anti-centromere which was significantly more prevalent in those without.
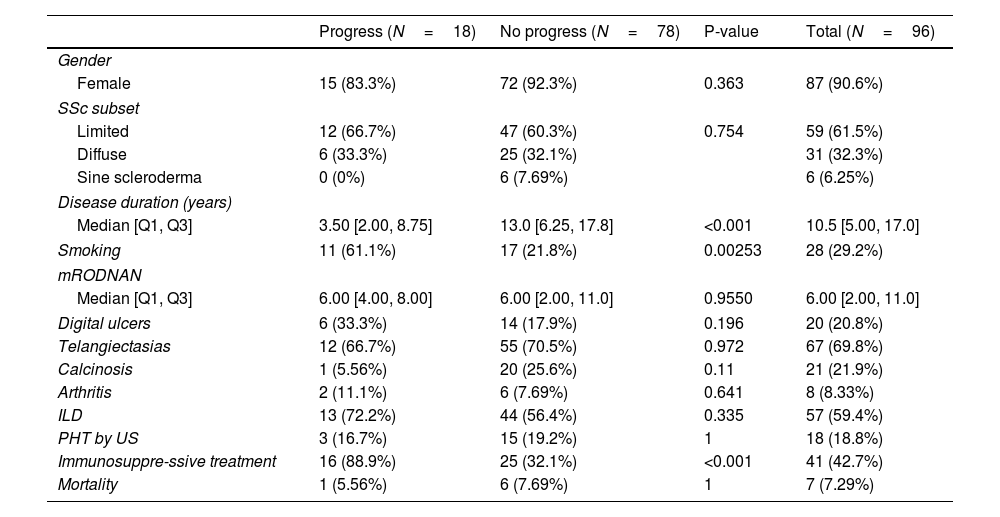
Table 3 shows a comparison between those patients who progressed regarding their ILD versus those who did not. Eighteen patients (19%) progressed. There were no significant differences between the groups regarding gender, subsets of SSc, clinical features or presence of autoantibodies. We did not find any association between degree of HRCT involvement and disease progression.
Comparison of patients whose ILD progressed and those who did not.
| Progress (N=18) | No progress (N=78) | P-value | Total (N=96) | |
|---|---|---|---|---|
| Gender | ||||
| Female | 15 (83.3%) | 72 (92.3%) | 0.363 | 87 (90.6%) |
| SSc subset | ||||
| Limited | 12 (66.7%) | 47 (60.3%) | 0.754 | 59 (61.5%) |
| Diffuse | 6 (33.3%) | 25 (32.1%) | 31 (32.3%) | |
| Sine scleroderma | 0 (0%) | 6 (7.69%) | 6 (6.25%) | |
| Disease duration (years) | ||||
| Median [Q1, Q3] | 3.50 [2.00, 8.75] | 13.0 [6.25, 17.8] | <0.001 | 10.5 [5.00, 17.0] |
| Smoking | 11 (61.1%) | 17 (21.8%) | 0.00253 | 28 (29.2%) |
| mRODNAN | ||||
| Median [Q1, Q3] | 6.00 [4.00, 8.00] | 6.00 [2.00, 11.0] | 0.9550 | 6.00 [2.00, 11.0] |
| Digital ulcers | 6 (33.3%) | 14 (17.9%) | 0.196 | 20 (20.8%) |
| Telangiectasias | 12 (66.7%) | 55 (70.5%) | 0.972 | 67 (69.8%) |
| Calcinosis | 1 (5.56%) | 20 (25.6%) | 0.11 | 21 (21.9%) |
| Arthritis | 2 (11.1%) | 6 (7.69%) | 0.641 | 8 (8.33%) |
| ILD | 13 (72.2%) | 44 (56.4%) | 0.335 | 57 (59.4%) |
| PHT by US | 3 (16.7%) | 15 (19.2%) | 1 | 18 (18.8%) |
| Immunosuppre-ssive treatment | 16 (88.9%) | 25 (32.1%) | <0.001 | 41 (42.7%) |
| Mortality | 1 (5.56%) | 6 (7.69%) | 1 | 7 (7.29%) |
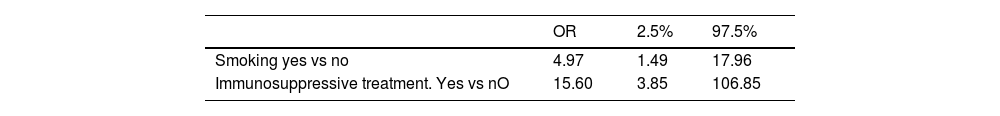
The only differences we found were a higher risk of progression with a history of smoking, having received immunosuppressive treatment and a shorter disease duration. Using a multivariate logistic regression model, these differences remained, with an OR of 4.97 (1.49–17.96) for a history of smoking and an OR of 15.6 (3.85–106.85) for having received immunosuppressive treatment (Table 4).
DiscussionOur purpose was to evaluate patients with SSc seen by both lung specialists and rheumatologists at a single University Hospital and study their characteristics particularly regarding interstitial lung disease. These patients were screened routinely at initial visit and did not necessarily have lung symptoms or signs at that time. As opposed to other research analyzing ILD and disease progression 23–25 these were not patients selected because they already had ILD.
Regarding SSc subsets, our patients showed a distribution between limited (61%) and diffuse (32%) in keeping with other reports5,14,15 and particularly the large series arising from EUSTAR with over 3000 patients.24 Clinical and serologic characteristics of both subsets did not differ from previous reports, in particular a higher mRSS and prevalence of anti Scl-70 in the diffuse patients.
The prevalence of ILD in this unselected population was 59%, also similar to other reports.14,15,24,25 Also as previously described, anti Scl-70 was significantly more prevalent in patients with ILD and inversely, anti-centromere significantly more prevalent in those without. In keeping with other series, the prevalence of more damage in HRCT was significantly higher in patients with ILD.
We did not find any significant differences in progression regarding clinical subsets, clinical features or antibody presence. In univariate analysis, disease duration was significantly shorter in those whose ILD progressed. In multivariate analysis the only significant differences were related to smoking status and the use of immunosuppressive treatment.
Regarding disease duration, it is well known that lung involvement preferably occurs within the first five years after onset of SSc so we could speculate that this is more related to this fact than other factors.26 A systematic review of papers evaluating ILD progression have also found a shorter disease duration associated with progression,27 as well as other series.27,28
Regarding immunosuppressive treatment, we may interpret that receiving this was related to the fact that patients with more extensive disease or who had shown progression were those being treated so this may explain this relationship. The OR was high but with a wide confidence interval. However, the minimum value was almost 4.
Other papers have mentioned the limitation of not being able to establish an association with immunosuppression, due to inadequate data because of the retrospective nature of these studies.28
As far as smoking is concerned, it has been analyzed in several reports without finding an association with ILD in SSc.29 We do not have an explanation for this finding.
We did not find an association of progression with autoantibodies,25 pulmonary hypertension, digital ulcers28 or degree of HRCT damage.30
The limitations of our work rely mainly on its retrospective design and the lack of control HRCT. These limitations have been emphasized as inherent to this type of research by several others.6,7,27,28
The strengths lie in the fact that this is a relatively large series for our country, followed by both rheumatologists and lung specialists with interest in this disease with adequate data regarding lung function tests over a relatively prolonged period.
It is clear from several of these reports25–30 that we are lacking a sufficiently useful measure to predict ILD progression in these patients which is essential for the management of this disease. For example, it has been shown that neither CVF decline on its own nor DLCO decline on its own are accurate enough. Periods of stability after decline of these variables and vice versa has been clearly demonstrated.26–28 A lack of association between PFT and imaging has also been reported.30
Finding adequate predictive factors will probably result from prospective trials designed to that effect.
In this local series, with clinical and serologic characteristics similar to those from other parts of the world, we only found that a shorter disease duration, smoking and the use of immunosuppressive therapy were associated with ILD disease progression.
Conflicts of interestsJIE has received fees from Boehringer Ingelheim for lectures and advisory boards.
The other authors have nothing to declare.
Boehringer Ingelheim (BI) provided an unrestricted educational grant to Fundación para el estudio de enfermedades fibrosantes del pulmón (FUNEF) for developing this study.
BI had no role in the design, analysis, or interpretation of the results in this study. BI was given the opportunity to review the manuscript for medical and scientific accuracy as well as intellectual property considerations.
We thank the Research Unit of the Argentine Society of Rheumatology (UNISAR) for their help in statistical analysis.
The authors would like to acknowledge the team of the ILD department of the University Hospital, José de San Martin and our patients.


