


La miositis por cuerpos de inclusión es una miopatía inflamatoria idiopática caracterizada por debilidad muscular, disfagia y biopsia muscular con inflamación y vacuolas ribeteadas. Presentamos el caso de una paciente a la que se le diagnosticó polimiositis (PM) pero que, por falta de respuesta al tratamiento, se le realizó una nueva biopsia que reveló miositis por cuerpos de inclusión.
Inclusion body myositis is a idiopathic inflammatory myopathy characterized by muscle weakness and dysphagia, with muscle biopsy showing inflammation and rimmed vacuoles. We present the case of a patient who was diagnosed with polymyositis but due to lack of response to treatment, a new biopsy revealed inclusion body myositis.
La miositis por cuerpos de inclusión (MCI) es una enfermedad inmunomediada que afecta los músculos y órganos internos1. Su prevalencia varía entre 24,8 y 45,6 por millón2. Afecta a individuos mayores de 50 años3. Cursa con daño musculoesquelético en la cara volar del antebrazo, flexores de los dedos y cuádriceps, causando debilidad muscular asimétrica y biopsia con cuerpos de inclusión4. Bioquímicamente cursan con elevación de la elevación de creatinfosfocinasa (CPK), presencia de anticuerpo anticitosólico 50-nucleotidasa cN1A hasta en un 30% de los casos, que a pesar de no ser específicos se asocian con mayor gravedad y mortalidad, y en la biopsia se observa un exudado inflamatorio endomisial con infiltración de células inflamatorias en fibras musculares no necróticas y vacuolas bordeadas por material citoplasmático membranoso, fibras atróficas e inclusiones congofílicas intra o extravacuolares3.
Presentamos a una paciente a la que se le diagnosticó polimiositis (PM) por elevación de CPK, anticuerpos AC-1 positivos, electromiografía (EMG) con patrón miopático y biopsia muscular con infiltrado inflamatorio, con poca respuesta al tratamiento esteroideo, por lo que se tomó una nueva biopsia que reveló MCI.
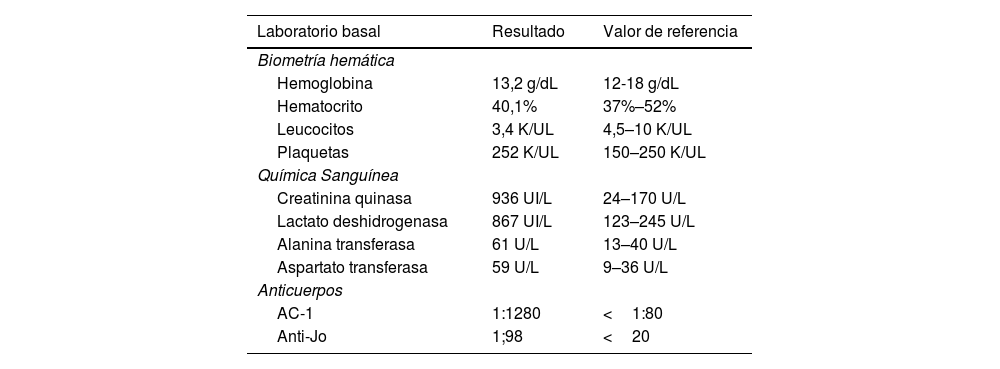
Presentación del casoMujer de 82 años de edad que a los 66 años presentó dificultad para subir escaleras y deglutir, disminución de fuerza muscular en cintura escapular y pélvica (4/5), elevación de CPK y anticuerpos AC-1 positivos. Posteriormente también se realizó inmunoespecificidad anti Jo1 y fue negativo (tabla 1). Se realizó EMG con patrón miopático y biopsia del músculo deltoides izquierdo bajo consentimiento informado con infiltrado inflamatorio endomisial linfocitario. Se le diagnosticó PM y se instauró tratamiento con prednisona y metotrexato, con posterior cambio a micofenolato mofetilo.
Laboratorios basales de paciente con miositis por cuerpos de inclusión
| Laboratorio basal | Resultado | Valor de referencia |
|---|---|---|
| Biometría hemática | ||
| Hemoglobina | 13,2 g/dL | 12-18 g/dL |
| Hematocrito | 40,1% | 37%–52% |
| Leucocitos | 3,4 K/UL | 4,5–10 K/UL |
| Plaquetas | 252 K/UL | 150–250 K/UL |
| Química Sanguínea | ||
| Creatinina quinasa | 936 UI/L | 24–170 U/L |
| Lactato deshidrogenasa | 867 UI/L | 123–245 U/L |
| Alanina transferasa | 61 U/L | 13–40 U/L |
| Aspartato transferasa | 59 U/L | 9–36 U/L |
| Anticuerpos | ||
| AC-1 | 1:1280 | <1:80 |
| Anti-Jo | 1;98 | <20 |
Evolucionó negativamente con uso de bastón y silla de ruedas, dificultad para extender los dedos de la mano izquierda, atrofia de músculos flexores largos de los dedos, predominantemente el izquierdo, y cuádriceps. De acuerdo con la evaluación muscular (procedimientos manuales de pruebas musculares MMT8/MMT26) se encontró una puntuación de 116 de 150 y 194 de 260, respectivamente, a expensas del involucro de los músculos distales. La nueva biopsia muscular mostró infiltrado inflamatorio con vacuolas ribeteadas y se diagnosticó MCI (fig. 1).
. Acercamiento 200× TMG, la tinción mostró fibras musculares de tamaño variable, abombamiento, agregados de células inflamatorias con invasión focal de algunas fibras musculares y una gran proliferación de tejido conectivo (A). Acercamiento a 400× TMG, con una evidente centralización nuclear (flecha) (B). Acercamiento 400× TMG Se observa vacuolas bordeadas con material granular y filamentos (estrella) clásicos de la patología (C). Tinción hematoxilina y eosina (H&E). Acercamiento 100×, vista panorámica de la biopsia donde se observa gran cambio en la arquitectura general, con aumento de los espacios y sustitución por tejido conectivo, morfología de las fibras con abombamiento e infiltrado inflamatorio (D), Acercamiento a 400× (H&E) muestra con más detalle el infiltrado inflamatorio que bordea a la fibra muscular (E) además de tejido conectivo abundante.")
Biopsia muscular del caso clínico.
Tinción tricrómico modificado de Gomori (TMG). Acercamiento 200× TMG, la tinción mostró fibras musculares de tamaño variable, abombamiento, agregados de células inflamatorias con invasión focal de algunas fibras musculares y una gran proliferación de tejido conectivo (A). Acercamiento a 400× TMG, con una evidente centralización nuclear (flecha) (B). Acercamiento 400× TMG Se observa vacuolas bordeadas con material granular y filamentos (estrella) clásicos de la patología (C).
Tinción hematoxilina y eosina (H&E). Acercamiento 100×, vista panorámica de la biopsia donde se observa gran cambio en la arquitectura general, con aumento de los espacios y sustitución por tejido conectivo, morfología de las fibras con abombamiento e infiltrado inflamatorio (D), Acercamiento a 400× (H&E) muestra con más detalle el infiltrado inflamatorio que bordea a la fibra muscular (E) además de tejido conectivo abundante.
Este caso se caracterizó inicialmente por debilidad muscular simétrica y proximal, disfagia y CPK elevada e infiltrado endomisial linfocitario, características de PM5. Sin embargo, la recurrencia de valores elevados de CPK, el aumento de la debilidad muscular proximal y la afección distal, la atrofia asimétrica de los músculos de las manos y cuádriceps, el empeoramiento de la disfagia y mala respuesta al tratamiento llevó a sospecha de MCI2. Alamr et al. informaron que el 14% de 367 pacientes con MCI tenían una presentación atípica y de estos el 6% de los pacientes comienzan con debilidad proximal del brazo6. En una serie de casos clínicos de MCI sin vacuolas ribeteadas en la biopsia, se retrasó el diagnóstico hasta ocho años después del inicio de los síntomas7; sin embargo, estas pueden estar ausentes en 20% de los casos8. El imitador más desafiante de MCI es la PM, ya que la serología es similar y los anticuerpos antinucleares (ANA) son positivos en 15-19% en MCI y 60% en PM9. Un estudio retrospectivo para comparar varios subtipos de miopatías inflamatorias no mostró una diferencia significativa entre PM y MCI en el seguimiento, la duración de la enfermedad, las características clínicas y los valores de CPK10. De hecho, actualmente la existencia de la PM se ha cuestionado, al observarse que la mayoría de los pacientes a quienes le fue diagnosticada PM, son reclasificados como síndrome antisintetasa sin exantema, miopatía necrosante inmunomediada o MCI con o sin vacuolas ribeteadas3,11.
El curso clínico de esta paciente deja ver el reto que es el diagnóstico diferencial entre la PM y MCI, lo que conlleva al retraso en el diagnóstico.
ConclusiónLa evolución clínica con afección muscular asimétrica y la mala respuesta a inmunosupresores, permitió replantear el diagnóstico hacia una MCI, con afección lentamente progresiva al igual que los cambios histopatológicos en las biopsias musculares. El tratamiento con corticoides e inmunosupresores no logró una detención de la progresión de la enfermedad.
FinanciaciónNo se requirió financiamiento externo. Se usaron los recursos del Hospital de Especialidades Centro Médico Nacional, La Raza.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


