




Se presenta el caso de una mujer de 50 años, fumadora, con artritis reumatoide seropositiva (FR y CCP) de 11 años de evolución en tratamiento con triple terapia, y aparición de nódulos pulmonares con diagnóstico final de histiocitosis de células de Langerhans por biopsia pulmonar. No hemos encontrado casos descritos de la coexistencia de ambas enfermedades. La abstinencia tabáquica llevó a la resolución radiológica sin necesidad de modificar la terapia inmunosupresora.
We report the case of a 50-year-old female smoker with an 11-year history of seropositive rheumatoid arthritis (rheumatoid factor and anti-cyclic citrullinated peptide antibodies) receiving triple therapy. She developed pulmonary nodules diagnosed as Langerhans cell histiocytosis by lung biopsy. We found no reported cases of the coexistence of these two diseases. Smoking abstinence led to radiologic resolution without modifying the immunosuppressive therapy.
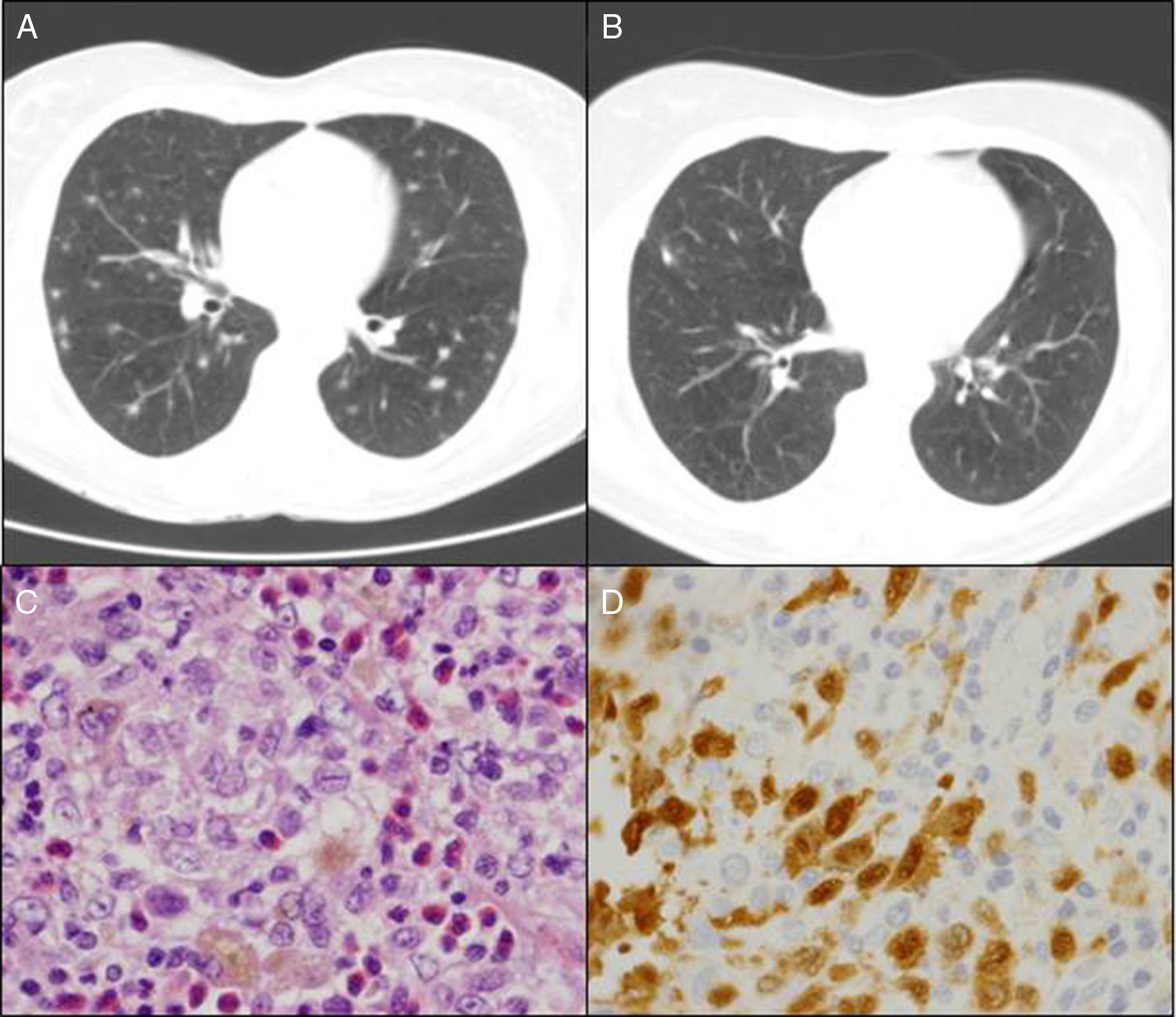
Mujer de 50 años, fumadora de 20 cigarrillos/día, con artritis reumatoide (AR) desde 1999, seropositiva (FR y CCP) y erosiva en tratamiento con metotrexato (MTX) desde el año 2000, y en combinación con salazopirina e hidroxicloroquina desde febrero 2009, con criterios de remisión completa desde entonces. En marzo del 2010, en relación con episodio catarral autolimitado, su médico de atención primaria había solicitado una radiografía de tórax donde se apreciaban dudosas imágenes nodulares de predominio en lóbulos superiores. La paciente se encontraba asintomática y la exploración física era normal. Destacaba un estudio inmunológico con FR y CCP positivos con ANA y ANCA negativos; Mantoux positivo, y un TAC torácico donde se observaban múltiples nódulos pulmonares bilaterales de unos 0,5cm, algunos con cavitación central de predominio en campos superiores y medios, pero también con afectación en ambas bases (fig. 1A). La broncoscopia resultó normal, con baciloscopia, cultivo del lavado broncoalveolar y citología de BAS para células malignas negativos. Fue derivada entonces para la realización de una biopsia pulmonar. En la videotoracoscopia se apreciaba un parénquima pulmonar con lesiones subpleurales de pequeño tamaño diseminadas, y en los resultados anatomopatológicos se apreciaba la presencia de histiocitos de citoplasma espumoso de núcleos con hendiduras, con otros multinucleados y algo elongados ocupando los espacios alveolares (fig. 1C). Las técnicas de inmunohistoquímica (IHQ) fueron positivas para CD1a (fig. 1D), S100, así como la langerina en las células histiocitarias descritas, compatible todo ello con histiocitosis de células de Langerhans (HCL). Se recomendó entonces abstinencia tabáquica absoluta y se mantuvo el mismo tratamiento inmunosupresor. Asintomática en todo ese periodo desde un punto de vista articular y respiratorio, se realizó control radiológico a los 6 meses, evidenciando imágenes quísticas pulmonares residuales y desaparición de los nódulos pulmonares (fig. 1B). Sin incidencias tras 5 años de seguimiento con mantenimiento de la triple terapia inmunosupresora.
Discusión TAC pulmonar al momento del diagnóstico. B) TAC pulmonar a los 6 meses del abandono del hábito tabáquico. C) Células de Langerhans (hematoxilina-eosina). D) Técnica IHQ positiva para CD1a (×100).")
Los histiocitos son células del sistema inmunitario que incluyen tanto a macrófagos como a las células dendríticas (no macrofágicas presentadoras de antígenos). Las histiocitosis son un grupo de enfermedades raras, siendo la HCL la entidad más representativa, caracterizada por la infiltración de células de Langerhans (CL), un tipo de célula dendrítica de localización predominante en el alvéolo pulmonar y en la piel, con sus característicos gránulos de Birbeck citoplasmáticos en forma de raqueta. El término de HCL fue acuñado con la intención de una mejor clasificación e identificación de los pacientes, ya que aglutinó a entidades anteriores (granuloma eosinófilo, histiocitosis X...) donde las lesiones se debían a una proliferación e infiltración del mismo tipo celular, siendo la identificación de la CL el criterio diagnóstico exigido desde entonces1. La patogénesis es desconocida, habiéndose encontrado una respuesta proliferativa tanto reactiva como clonal con distinto grado de agresividad fenotípica en los órganos o sistemas infiltrados (tabla 1). La histiocitosis de células de Langerhans pulmonar (HCLP) es la forma más representativa del adulto y suele reconocerse como una entidad independiente.
Clasificación de la histiocitosis
| Clasificación simplificada de las histiocitosisa |
|---|
| 1- Langerhans cell histiocytosis (LCH) |
| 2- Hemophagocytic lymphohistiocytosis (HLH) |
| 3- The rare histiocytic disorders (RHD) |
| Juvenil xanthogranuloma |
| Erdheim-Chester disease |
| Multifocal reticulohistiocytosis |
| Rosai-Dorfman disease |
| The malignant histiocytosis |
| Clasificación de las histiocitosis de células de Langerhans |
|---|
| 1- Enfermedad aislada de un único órgano o sistemab |
| HCL pulmonar (85% de los casos de las HCL pulmonares del adulto) |
| Ósea (única o múltiple) |
| Piel/hipotálamo/hipófisis/ganglios linfáticos/hígado, bazo, tiroides |
| 2- Enfermedad multisistémicac: afectación de dos o más órganos |
La HCLP se produce en adultos jóvenes fumadores, y la abstinencia tabáquica puede conducir a la remisión parcial o total de las lesiones pulmonares. Se caracteriza en las fases iniciales por cambios inflamatorios bronquioloalveolares y en las fases más avanzadas, por destrucción quística pulmonar. Las manifestaciones clínicas son muy variadas: disnea, tos, astenia, fiebre, pérdida de peso, dolor torácico pleural y, en ocasiones, neumotórax espontáneo. La incidencia de hemoptisis es anecdótica, por lo que si esta se produce, deben buscarse otras causas de la misma (principalmente tumoral). La enfermedad se puede descubrir radiológicamente de manera casual en pacientes asintomáticos en alrededor del 15% de los casos, y en un mismo porcentaje pueden aparecer manifestaciones extrapulmonares como quistes óseos, diabetes insípida o exantema2,3. La tomografía axial computarizada se prefiere para caracterizar mejor el compromiso pulmonar, y la combinación de múltiples quistes y nódulos bilaterales distribuidos en campos medios y superiores, con o sin engrosamiento intersticial, en un joven fumador se considera característico y de alta sospecha diagnóstica4, situación que no sucedía en nuestra paciente, dado que presentaba patrón principalmente nodular, escasas lesiones quísticas y con afectación de lóbulos inferiores. El lavado broncoalveolar es útil para el diagnóstico cuando más de un 5% de las CL son detectadas por microscopia electrónica o inmunotinción, pero es una técnica no disponible en todos los centros hospitalarios5. La combinación de los hallazgos radiológicos típicos junto con la positividad del lavado broncoalveolar puede ser aceptada como criterio diagnóstico suficiente, sin necesidad de confirmación anatomopatológica2, pero si esta fuese necesaria, debe realizarse una biopsia pulmonar dado que la biopsia transbronquial ha demostrado un rendimiento diagnóstico muy reducido. Las CL se confirman por técnicas inmunohistoquímicas de anticuerpos monoclonales contra el antígeno de membrana CD1a, la proteína intracelular S100 y la langerina6.
El diagnóstico diferencial debe incluir principalmente el cribado protocolario de micobacterias, infecciones con diseminación hematógena, otras nodulosis como sarcoidosis, silicosis, vasculitis, metástasis o tumores primarios pulmonares, y en nuestro caso obviamente con nódulos reumatoideos (NR) pulmonares. Respecto a estos últimos, recordar que los NR pulmonares se relacionan clásicamente con el tabaquismo, la positividad del FR, la coexistencia de NR cutáneos, el HLA DRB1, pero también existen los casos inducidos por el propio tratamiento con FAME sintéticos o la terapia biológica anti-TNF. La nodulosis reumatoidea inducida por MTX es una entidad hace ya tiempo aceptada,y que se caracteriza por el rápido desarrollo de NR histopatológicamente similares a los NR clásicos, que ceden tras la suspensión del mismo y reaparecen nuevamente cuando se intenta volver a reintroducir7. Se ha asociado con alelo HLA DRB1*0401. La nodulosis reumatoidea pulmonar de similares características cronológicas inducida por MTX es mucho menos frecuente. Por el contrario, otras situaciones en que se describe la aparición de NR cutáneos o pulmonares durante el transcurso de un tratamiento crónico y prolongado con MTX, la asociación causal sería mucho más cuestionable, y además es característica en la mayoría de las ocasiones la discordancia entre el desarrollo de la nodulosis y un buen control de la enfermedad articular. Es discutible también entonces la actitud a seguir respecto a la retirada del fármaco, y todo ello lleva a pensar en mecanismos fisiopatológicos diferentes entre los que llevan a la formación del granuloma reumatoideo y los que conducen a la sinovitis e hipertrofia sinovial. Problemas de interpretación similares en la literatura nos vamos a encontrar con la leflunomida y la terapia anti-TNF como inductores de nodulosis reumatoidea pulmonar8. Finalmente, el diagnóstico diferencial de la forma quística pura de la HCLP incluiría el enfisema centrolobulillar, la fibrosis quística, la esclerosis tuberosa y, en mujeres específicamente, la linfangioleiomiomatosis (tabla 2).
Diagnóstico diferencial nódulos pulmonares en paciente con HCL pulmonar y AR
| Tumoral | |
| Cáncer de pulmón | |
| Metástasis | |
| Carcinomatosis linfangítica | |
| Infeccioso | |
| Infección hematógena: Staphylococcus aureus, Klebsiella pneumoniae, Pseudomonas | |
| Granulomatosis: micobacterias, Nocardia, hongos | |
| Parásitos: hidatidosis, paragonomiasis, Echinococcus | |
| Inflamatorio | |
| Sarcoidosis | |
| HCL pulmonar | |
| Neumoconiosis: asbestosis, silicosis, síndrome Caplan | |
| Nódulos reumatoideos | |
| Vasculitis: granulomatosis con poliangeítis, granulomatosis eosinofílica | |
| Neumonía criptogénica organizada | |
| Fármacos | |
| Nódulos reumatoideos inducidos por FAME (MTX y LEF) y anti-TNF | |
| Amiodarona, bleomicina, carbamazepina, otros | |
| Miscelánea | Diagnóstico diferencial del patrón quístico radiológico |
| Bronquiolitis respiratoria | HCL pulmonar |
| Amiloidosis | Linfangioleiomiomatosis |
| Embolismo pulmonar | Fibrosis quística |
| Malformación congénita de la vía aérea | Esclerosis tuberosa |
El curso natural de la HCLP es impredecible, estimándose hasta en un 50% de los casos las resoluciones espontáneas, y en un 10-20% la progresión con insuficiencia respiratoria progresiva severa. El tratamiento principal de la HCL pulmonar es la abstinencia tabáquica absoluta, que puede acompañarse de la resolución hasta en una tercera parte de los casos, y simultáneamente se elimina un importante factor de riesgo de cáncer de pulmón, EPOC y enfermedad cardiovascular. Sin embargo, se estima que hasta en un tercio de los pacientes que dejan de fumar la enfermedad puede continuar progresando. También se han descrito casos de recidiva en pacientes con abstinencia tabáquica absoluta. El uso de corticoides en monoterapia o en combinación con inmunosupresores (metotrexato, ciclofosfamida, vinblastina y más recientemente con cladribina) está cuestionado por la ausencia de ensayos controlados y por la incertidumbre del propio comportamiento natural de la enfermedad. Suele reservarse en casos de deterioro funcional espirométrico, enfermedad progresiva intersticial o en la afectación multisistémica extrapulmonar. La supervivencia global a los 5 años se estima del 75%, y los factores de mal pronóstico más aceptados son la edad avanzada y el deterioro en las pruebas de función respiratoria y difusión al momento del diagnóstico3. El trasplante de pulmón es una opción en enfermedad severa evolucionada, aunque se han descrito recidivas sobre el órgano trasplantado9.
Nuestra paciente estaba en remisión clínica articular recibiendo tratamiento con triple terapia cuando se detectaron las manifestaciones pulmonares, y mantuvimos la misma estrategia principalmente al comprobar la mejoría radiológica posterior tras la abstinencia tabáquica. No hemos encontrado casos comunicados en la literatura de la coincidencia previa de HCLP y AR. Existe recientemente descrito un caso de HCLP en un paciente con síndrome de Sjögren tratado con esteroides orales y azatioprina, con buena respuesta radiológica y funcional10. La reticulohistiocitosis multifocal puede ser una causa de artritis erosiva y se ha descrito asociada en pacientes con AR y Sjögren11, aunque clínicamente es un diagnóstico diferencial no considerado en nuestro caso. Respecto a la actitud a tomar si en algún momento precisase de terapia biológica, únicamente hemos podido rescatar la comunicación favorable de Henter et al., en el año 2001, de un paciente con HCL infantil multifocal muy agresiva con buena respuesta a etanercept12, pero desde entonces no se han repetido comunicaciones en la misma línea.
En resumen, la HCLP es el tipo más representativo de HCL en la edad adulta. Se relaciona directamente con el hábito tabáquico, y la abstinencia del mismo conduce en un alto porcentaje de los casos a la resolución de la enfermedad. No hemos encontrado referencias previas en la literatura de la coexistencia simultánea de AR y HCLP. Nuestro caso no presentaba el patrón radiológico característico, y el tratamiento inmunosupresor con triple terapia no parece haber influido en la evolución sintomática favorable después de más de 5 años de seguimiento tras la supresión tabáquica.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Conflicto de interesesLos autores declaran no tener conflictos de intereses.


