


La interacción molecular doble y simultánea entre las células presentadoras de antígeno (CPA) y los linfocitos T es imprescindible para la activación óptima de la respuesta inmunitaria y requiere de la participación de dos grupos de receptores de membrana. El abatacept es una proteína de fusión que modula selectivamente una de estas dos vías, uniéndose a los receptores CD80 y CD86 de las CPA. De esta forma el fármaco inhibe la activación de las células T, bloqueando selectivamente la unión específica de los receptores CD80/CD86 al CD28 y como consecuencia inhibiendo la proliferación de las células T y la respuesta inmunitaria de las células B. Esta acción farmacológica se traduce en la normalización de los niveles de los mediadores inflamatorios en los enfermos con artritis reumatoide y en una respuesta clínica segura y eficaz. El abatacept, en combinación con metotrexato, evita la progresión de la lesión articular y mejora la función física en enfermos con artritis reumatoide.
The double and simultaneous molecular interaction between antigen-presentig cells (APC) and T lymphocytes is essential for the optimal activation of the immunological response and requires the participation of two membrane receptor groups. Abatacept is a fusion protein that selectively modulates one of these two ways, by binding to CD80 and CD86 receptors on APC. In this way, the drug inhibits T cell activation, selectively blocking the specific interaction of CD80/CD86 receptors to CD28 and, therefore, inhibiting T cell proliferation and B cell immunological response. This pharmacological action results in the normalization of inflammatory mediators in rheumatoid arthritis patients and in a safe and efficacious clinical response. Abatacept in combination with methotrexate prevents the progression of joint damage and improves physical function in rheumatoid arthritis patients.
La artritis reumatoide (AR) es una enfermedad autoinmune, de carácter poligénico, caracterizada por una poliartritis con manifestaciones sistémicas y una elevada y grave morbilidad1,2. La AR afecta del 0,5 al 1% de la población, ocasionando una disminución de la calidad de vida, importante discapacidad física y un cuantioso coste económico3–6. La expresión clínica de la enfermedad es muy variada, abarcando desde formas autolimitadas leves a otras muy agresivas, que experimentan una rápida evolución que culmina con la destrucción de la articulación afectada y la consiguiente limitación funcional7.
Los estudios genéticos han confirmado la existencia de un sustrato genético, asociado en parte a determinados genes codificadores de proteínas que participan en la respuesta de los linfocitos T1. Estos hallazgos refuerzan la importancia del papel atribuido a las células T en el inicio y perpetuación anómala de la respuesta inmune en esta enfermedad8.
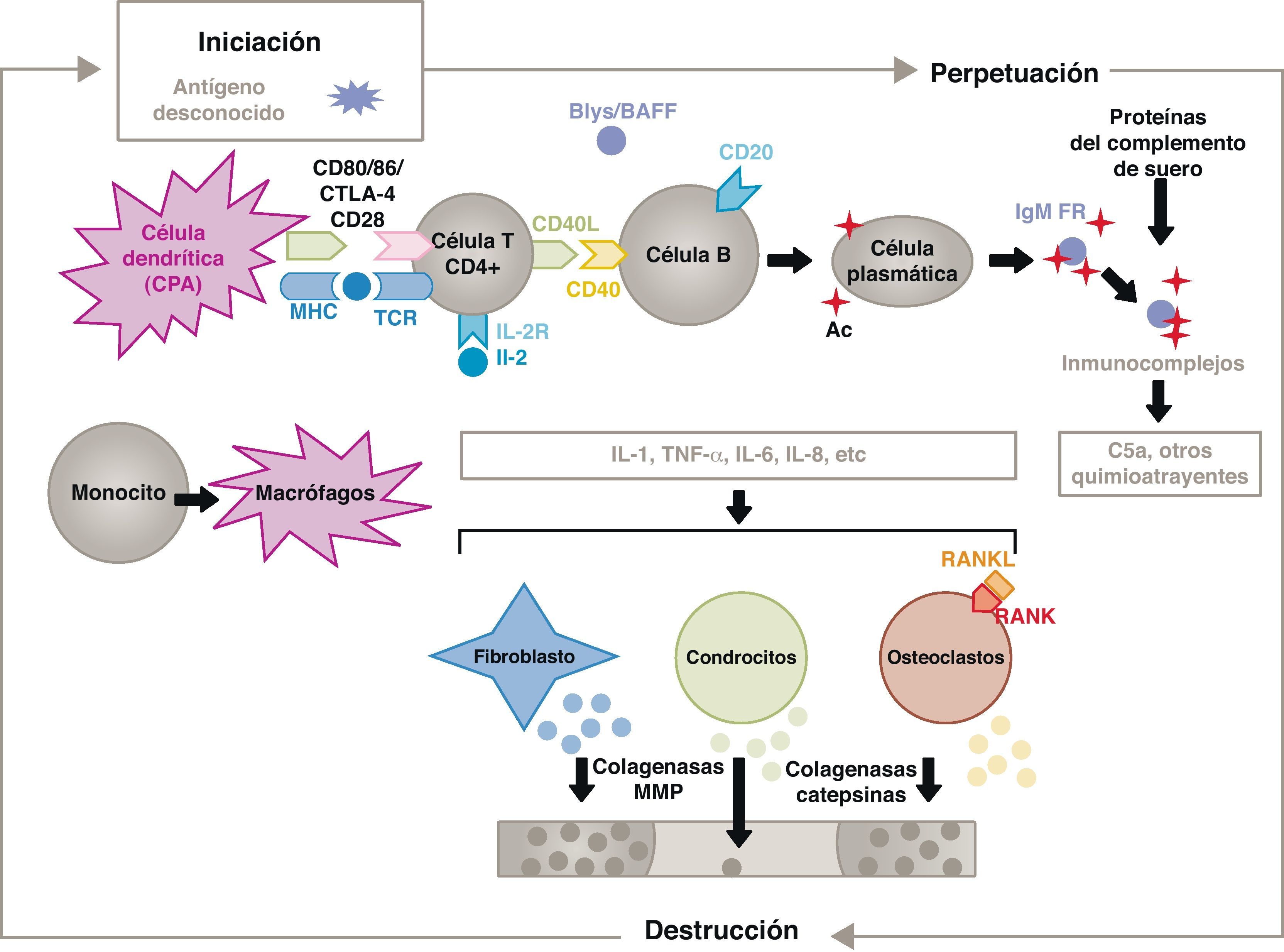
La patogenia de la AR es compleja y en ella intervienen diferentes poblaciones celulares implicadas en la respuesta inmune innata y adquirida. En su patogenia participan células residentes en la membrana sinovial, como los sinoviocitos B de estirpe fibroblástica o los macrófagos de la íntima, y las células inflamatorias provenientes de la sangre como los linfocitos T, los linfocitos B y los monocitos9. Todas ellas contribuyen a la transformación agresiva del fenotipo de los sinoviocitos B y al desarrollo de un intenso infiltrado inflamatorio cuyo resultado final es la destrucción del cartílago y del hueso subcondral10,11 (fig. 1).

Fisiopatología de la artritis reumatoide. Esquema fisiopatológico general de la artritis reumatoide. AC: anticuerpo; BAFF: factor activador de células B; Blys: estimulador de linfocitos B; CD: cúmulo de diferenciación; CPA: célula presentadora de antígeno; CPH: complejo principal de histocompatibilidad; CTLA4: antígeno 4 asociado al linfocito T citotóxico; C5a: fracción 5a del complemento; FR: factor reumatoide; Ig: inmunoglobulina; IL: interleucina; MMP: metaloproteasas de la matriz; RANK: receptor activador del factor nuclear κappa B; RANKL: ligando de receptor activador para el factor nuclear κappa B; RCT: receptor de linfocitos T; TNF: factor de necrosis tumoral.
El tratamiento actual de la AR se basa en la administración de fármacos antirreumáticos modificadores de la enfermedad (FAME) utilizados en monoterapia o en combinación12. Estos fármacos retrasan la destrucción articular, es decir, son capaces de modificar el curso natural de la enfermedad4,13. No obstante, el porcentaje de enfermos con una respuesta clínica satisfactoria es bajo y a menudo obliga a la adición de un FAME biológico en un elevado porcentaje de pacientes9,13–15.
En los últimos años se han identificado nuevas moléculas y dianas terapéuticas cuyo bloqueo podría aminorar o suprimir la respuesta inflamatoria crónica. Una de estas nuevas moléculas es abatacept. El abatacept es un constructo proteico, totalmente humanizado, formado por el dominio extracelular del antígeno 4 asociado al linfocito-T citotóxico humano (CTL4) y un fragmento genéticamente modificado de la región Fc de la inmunoglobulina humana G1 (IgG1), que inhibe la coestimulación de las células T actuando sobre el verdadero núcleo inicial de la respuesta inmunitaria y, por tanto, en el inicio de la enfermedad.
Activación de las células TLa activación inmunitaria eficaz de las células T requiere de la participación de dos grupos de receptores de membrana en las células presentadoras de antígeno (CPA)14 (figs. 1 y 2). El primero es el vehículo que utilizan las CPA para ofrecer a la célula T un antígeno específico previamente procesado por la célula. A pesar del enorme esfuerzo dedicado a esta investigación, todavía no se ha podido identificar el/los antígenos artritogénicos desencadenantes de la AR8. La presentación por la CPA de un antígeno, frente al que desencadenar una respuesta inmunitaria específica, al linfocito T se organiza a través de un complejo trimolecular que conforman: moléculas del complejo principal de histocompatibilidad (CPH) presentes en las CPA, el antígeno frente al que se desarrolla la respuesta inmunitaria y un receptor de membrana en la célula T (RCT) específico para ese antígeno15 (señal o vía de señalización 1 de la respuesta inmunitaria).

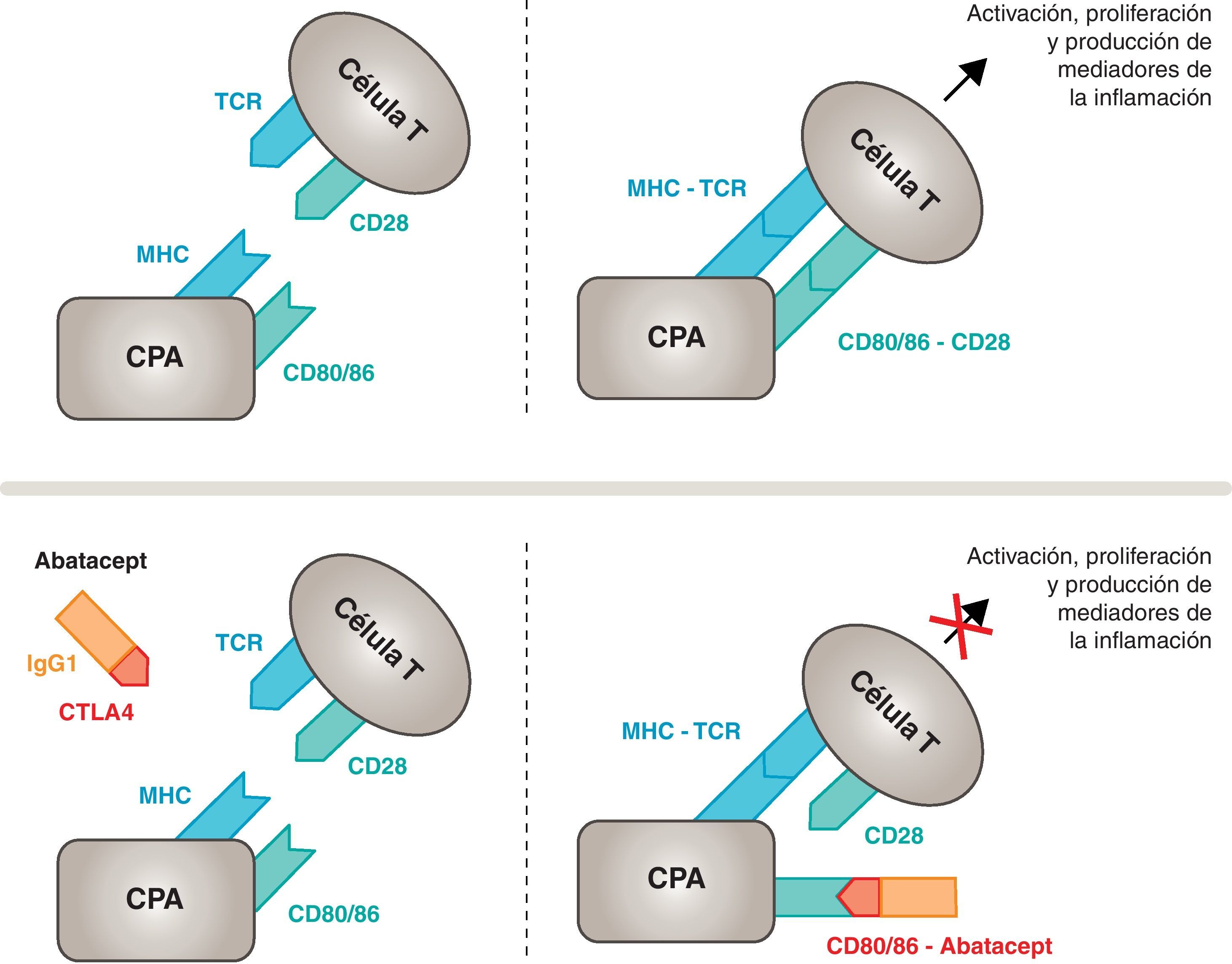
Mecanismo de acción del abatacept. El fragmento del abatacept constituido por el dominio extracelular del CTLA4 se une a los receptores CD80/CD86, evitando o desplazando su interacción con el receptor CD28. De esta forma se bloquea selectivamente la unión específica de los receptores CD80/CD86 al CD28, lo que equivale, fisiopatológicamente, a bloquear la segunda señal de la activación inmunitaria y, por tanto, la activación de las células T. CPA: célula presentadora de antígeno; CPH: complejo principal de histocompatibilidad; RCT: receptor de células T.
Para promover la activación completa de la célula T es necesario un segundo bloque de comunicación intercelular entre las CPA y los linfocitos T, que se produce a través de las vías coestimuladoras y constituyen la denominada señal 2 de la respuesta inmunitaria14. Aunque existen varias vías coestimuladoras, una fundamental es la que resulta de la unión de los receptores CD80 (B7-1)/CD86 (B7-2) en la membrana de las CPA y el receptor CD28 en los linfocitos T10,16. La activación simultánea de ambas señales, 1 y 2, desencadena una intensa señalización intracelular en los linfocitos T, imprescindible para su activación completa, proliferación, supervivencia y producción de citocinas8. A las 24-48 h de producirse la activación de los linfocitos T, la misma señalización intracelular inicia un mecanismo regulador que tiene como objetivo la desactivación de la propia respuesta. Así se induce la expresión en la membrana celular linfocitaria del CTLA411, con la misión de competir con el CD28 dada su mayor afinidad de unión al CD80/CD8617,18.
La activación de los dos subgrupos de células T, CD4+ y CD8+ depende de la coestimulación del receptor CD28. Las células T CD4+ son las células T colaboradoras. Éstas, reconocen los péptidos ofrecidos por las moléculas de la clase II del CPH presentes en las CPA. Estos antígenos se originan a partir de la vía exógena que procesa patógenos como las bacterias. Muchas enfermedades autoinmunes se asocian a una respuesta patológica de las células T CD4+.
Por su parte, las células T CD8+ son los linfocitos citotóxicos (LCT). Los receptores de las células T CD8+ reconocen antígenos, principalmente virales y tumorales, presentados por moléculas de clase I del CPH. Tras su activación, las células CD8+ median la destrucción de las células diana mediante la producción de perforina, granzimas e interferón (IFN)-γ.
Ambos subtipos de células T son activados mediante la coestimulación con CD2815, aunque la activación de las células T CD8+ es menos dependiente de esta vía de coestimulación. De hecho, mientras todas las células CD4+ expresan el receptor CD28 en su membrana, esto sólo ocurre en alrededor del 50% de las CD8+19. Además, las células CD4+ han demostrado exhibir una mayor respuesta a la unión a CD2820. Por otra parte, el activador CD28 no es un requisito absoluto para la activación de LCT21. Todo lo cual aportaría un doble beneficio terapéutico en la práctica clínica. Por un lado, abatacept actúa preferentemente sobre la célula diana de la patogenia de la enfermedad. Por otro, la acción reducida sobre la actividad de los linfocitos CD8+ aseguraría un mejor perfil de seguridad en cuanto a complicaciones virales y tumorales.
La activación de las células T CD4+ es el punto de partida de una cascada de fenómenos proinflamatorios con producción de gran cantidad de citocinas y proliferación celular que, en caso de perpetuarse de forma mantenida, como ocurre en la AR, da lugar a una inflamación crónica muy activa, capaz de destruir los tejidos en los que se desencadena, principalmente en las articulaciones en el caso de la AR8 (fig. 1). En la membrana sinovial comienzan a proliferar las células infiltrantes provenientes de la sangre, como los propios linfocitos T y sus subtipos, y también los linfocitos B. Los monocitos se diferencian en macrófagos y osteoclastos y, además, activan a los condrocitos articulares. En este medio se producen grandes cantidades de citocinas proinflamatorias como la interleucina (IL)-1, la IL-6 y el factor de necrosis tumoral (TNF) entre otras muchas. Las células B producen también autoanticuerpos como el factor reumatoide o los anticuerpos antipéptidos citrulinados. Todo lo cual conduce a la destrucción no sólo de la membrana sinovial, sino también del hueso subyacente y del cartílago articular22.
La biotecnología en el tratamiento de la artritis reumatoideComo consecuencia de las investigaciones previamente mencionadas, se han ido desarrollando y comercializando varias moléculas de fabricación biotecnológica dirigidas a bloquear dianas específicas. En una primera generación aparecieron los fármacos neutralizantes del TNF: etanercept, infliximab y adalimumab y otro, el anakinra, para inhibir la acción de la IL-1. Posteriormente, han surgido nuevas moléculas, como el abatacept, para modular la coestimulación de la respuesta inmunitaria; el certolizumab y el golimumab para bloquear el TNF; el rituximab contra el receptor CD20 de los linfocitos B, y el tocilizumab frente al receptor de la IL-67,23–26.
A pesar del enorme salto cualitativo en lo que respecta a la eficacia terapéutica que ha supuesto la introducción de estos fármacos, un porcentaje sustancial de enfermos, estimado entre el 25 y 40%, no responde a los fármacos biológicos actualmente comercializados o deben ser interrumpidos por la aparición de acontecimientos adversos27–32. La necesidad de mejorar esta situación mantiene el estímulo en la búsqueda y el desarrollo de nuevas moléculas dirigidas a regular o tratar dianas terapéuticas diferentes que puedan mejorar la eficacia terapéutica como es el caso de abatacept, que modula selectivamente la activación de las células T33.
El abatacept es un constructo proteico que se obtiene mediante tecnología de ADN recombinante en células de ovario de hámster34,35. Esta molécula fue diseñada para interferir con la regulación de las vías de coestimulación de las células T, que tienen un papel importante en la patogenia de diversas enfermedades autoinmunes, infecciones, rechazo de órganos trasplantados e inmunidad tumoral36.
El abatacept se utiliza en combinación con metotrexato en pacientes con AR que hayan presentado una respuesta inadecuada o intolerancia a otros FAME, incluidos metotrexato (MTX) o un inhibidor del TNF-alfa. En artritis idiopática juvenil poliarticular está indicado en pacientes pediátricos de 6 o más años con una respuesta inadecuada a otros FAME, incluidos al menos un neutralizante del TNF35.
Mecanismo de acción del abataceptEl abatacept es un modulador selectivo de la señal coestimuladora CD80/86:CD28 que, como se ha comentado previamente, es imprescindible para la activación de las células T. El abatacept inhibe la activación de las células T, bloqueando selectivamente la unión específica de los receptores CD80/CD86 de las CPA al CD28 del linfocito T (fig. 2)22,37. La estrategia farmacológica buscaría desactivar la respuesta inmuno/inflamatoria acelerada, característica de la enfermedad, y reponer la homeostasis normal en el sistema inmune. De hecho, la competencia entre el CD28 y el CTLA4 endógeno por la unión al CD80/86 es el mecanismo fisiológico que permite regular y, en su caso, concluir con la respuesta inmunológica normal. Abatacept, al bloquear la unión de CD80/86 al CD28 inhibe la transmisión de la segunda señal de la respuesta inmunitaria, lo que indirectamente produce una señal negativa sobre la activación de las células T. Además, abatacept tiene probablemente un mayor efecto en prevenir la formación de la señal coestimuladora que en desactivar a la célula T ya activa, ya que no está unido al CTLA4 de las células T.
Respaldo farmacológico para su uso1. ¿Por qué se incluye en el grupo de fármacos inmunorreguladores? Fundamentalmente, porque no produce depleción celular, especialmente de células T sobre las que ejerce la acción farmacológica ni tampoco bloquea selectivamente una citocina particular, evitando la supresión radical de efectores esenciales para un adecuado funcionamiento de la respuesta inmunitaria8.
2. ¿Cómo se ha evitado la unión de la molécula a los receptores Fc? La región Fc del abatacept está genéticamente modificada, de forma que no se une a los receptores CD16 y CD32, y lo hace muy débilmente, a los receptores CD64. Este diseño permite soslayar las respuestas celulares mediadas por el receptor Fc como son la citotoxicidad celular dependiente de anticuerpo (CCDA) y la citotoxicidad dependiente del complemento (CDC)18. Ambas se asocian a lisis celular, con los potenciales efectos adversos que puedan ocasionar en tratamientos prolongados38. Por tanto, el fragmento modificado de la IgG1 no parece ser activo con lo que evitaría los acontecimientos adversos resultantes de la CCDA y la CDC39.
3. Efecto antiinflamatorio del abatacept. El abatacept reduce de forma significativa muchos de los mediadores inflamatorios en pacientes con AR, restituyéndolos a la normalidad; hecho demostrado en varios ensayos clínicos utilizados para el registro del fármaco.
En un estudio en fase II-b, de 1 año de duración, controlado con placebo, en pacientes con AR y respuesta inadecuada al MTX, se obtuvieron muestras en los días previos a la infusión y se determinaron niveles séricos de marcadores seleccionados con el objetivo de estudiar el efecto del abatacept en los mediadores y citocinas proinflamatorias. Un grupo de enfermos recibió MTX y abatacept 10mg/kg, según pauta habitual. El grupo control, por su parte, fue tratado con MTX y placebo. Al año de tratamiento, los marcadores biológicos en el grupo de abatacept 10mg/kg se habían normalizado, mientras que los del grupo placebo seguían elevados (TNF: 7,4 vs. 10,3pg/ml; FR: 159 vs. 225 U/l; sIL-2R: 1.228,3 vs. 1.697,1pg/ml; IL-6: 7,3 vs. 19,9pg/ml)40.
4. Inmunogenicidad. Según datos procedentes del registro del fármaco, sólo 187 de 3.877 (4,8%) pacientes con AR tratados durante períodos de hasta 8 años con abatacept desarrollaron anticuerpos frente al fármaco durante el tratamiento41. En los pacientes en los que se evaluaron los anticuerpos frente a abatacept después de interrumpir el fármaco (> 42 días después de la última dosis), 103 de 1.888 (5,5%) fueron seropositivos. Por el contrario, en otro estudio en 2.000 pacientes no se detectaron anticuerpos contra abatacept. Ciertamente, el abatacept presenta un bajo nivel inmunogénico42,43.
5. Abatacept y tuberculosis. El TNF participa de forma destacada en la respuesta inflamatoria e inmunopatológica de la tuberculosis (TB). Los estudios in vitro muestran que el TNF aumenta la actividad fagocitaria y micobactericida de los macrófagos, mientras que in vivo interviene en la fase inicial de formación y el posterior mantenimiento de los granulomas, formaciones que controlan el crecimiento de las micobacterias y limitan su propagación. En un modelo crónico de reactivación de TB latente en ratones, se ha estudiado la evolución de la infección en ratones tratados con abatacept frente a otro grupo tratado con un monoclonal anti-TNF murino42. A los 4 meses de infectar los ratones C57BL/6 con Mycobacterium tuberculosis y, una vez confirmada la TB latente, los ratones recibieron tratamiento durante 16 semanas con una de las dos intervenciones experimentales. Al cabo de este tiempo, todos los ratones tratados con el anti-TNF habían muerto por una TB diseminada con una supervivencia media de 44 días. Por el contrario, no murió ninguno de los ratones tratados con abatacept.
Mientras que la concentración de IFN-γ en suero no se modificó en el grupo con abatacept, ésta se encontró elevada en los ratones con anti-TNF. Dicha elevación fue atribuida a la mayor infiltración de células CD4+ y CD8+ originada por la dispersión generalizada del número de colonias bacterianas.
Así pues, mientras que los ratones tratados con terapia anti-TNF mostraron una mortalidad del 100%, el abatacept no alteró la capacidad de los ratones para organizar una respuesta inflamatoria capaz de controlar la diseminación tuberculosa. No obstante, todavía no se dispone de datos clínicos suficientes que confirmen estos mismos hallazgos en humanos.
6. Efecto antirresortivo del abatacept en el remodelado óseo. La actividad osteoclástica está incrementada en la AR, tanto en la articulación, provocando erosiones óseas, como a nivel sistémico, asociada a una osteoporosis generalizada44,45.
De hecho, se ha demostrado un incremento del ligando del receptor activador del factor nuclear NF-kB (RANKL) en la membrana sinovial45,46. El abatacept inhibe de forma dependiente de la dosis la formación de osteoclastos murinos, así como la actividad osteoclastogénica valorada in vitro. Esta fue estudiada en cultivos de osteoclastos murinos sobre placas de dentina, en las que se midió el número de hoyos de resorción a los 6 días de añadir diferentes dosis del abatacept47. El fármaco disminuyó significativamente el área de resorción ósea.
Estos datos sugieren que abatacept es una molécula antiosteoclastogénica que se une directamente a las células precursoras del osteoclasto, inhibiendo su diferenciación. Este mecanismo podría explicar el efecto anti-erosivo del fármaco en enfermos con AR. De hecho, los enfermos tratados con abatacept muestran una tendencia a la disminución del RANK, así como de su ligando RANKL en la membrana sinovial, todo ello asociado a un aumento de la osteoprotegerina48. Aunque no está claro el mecanismo exacto subyacente a esta observación, estos hallazgos se correlacionan bien con la mejoría radiológica observada en enfermos tratados con abatacept.
7. Efecto del abatacept en otras células inmunitarias. A pesar de que la CPA es la célula diana a la que se une el abatacept y que el macrófago expresa también en su superficie receptores CD80/86, existen pocos estudios que investigan la acción del fármaco en la actividad de estas células. En efecto, un estudio reciente in vitro ha demostrado que los macrófagos exhiben una marcada expresión de receptores CD80/86 y que su tratamiento con abatacept reduce sustancialmente la producción de citocinas49. Estos resultados sugieren que el mecanismo de acción del fármaco podría ampliarse a la regulación de la estirpe macrofágica, célula clave en la patogenia de la enfermedad.
El abatacept suprime también la migración folicular de las células T específicas de antígeno y, consecuentemente, la colaboración entre la célula T folicular y la célula B en el ganglio linfático. Este hallazgo se ha observado in situ en ganglios linfáticos de ratones BALB/c50. Tras transfundir a dichos ratones linfocitos T preestimulados con un antígeno específico, una posterior inmunización de los ratones mostró proliferación de las células T y su migración al área linfática B. En los ratones tratados con abatacept se bloqueó la proliferación de las células T y también su migración, limitándose su presencia, en la mayoría de los casos, a la zona paracortical ganglionar. En consecuencia, el tratamiento prolongado con abatacept disminuiría la proliferación, la movilidad y la distribución intraganglionar de los linfocitos con memoria de autoantígeno, lo cual podría dar lugar al descenso de autoanticuerpos.
Conclusiones sobre el mecanismo de acción de abataceptEl abatacept es un constructo proteico, totalmente humanizado, formado por el dominio extracelular del antígeno 4 asociado al linfocito-T citotóxico humano (CTL4) y un fragmento genéticamente modificado de la región Fc de la IgG1, diseñado para interferir con la regulación de la vía de coestimulación de los linfocitos T. El fármaco inhibe la activación de las células T, bloqueando selectivamente la unión específica de los receptores CD80/CD86 al CD28 y, como consecuencia, inhibiendo la proliferación de las células T y de la respuesta inmunitaria de los linfocitos B. Esta acción farmacológica se traduce en la disminución de los niveles de los mediadores inflamatorios en los enfermos con AR y en una respuesta clínica segura y eficaz.
Conflicto de interesesEl Dr. Gabriel Herrero-Beaumont ha recibido ayudas para investigación de Bristol-Myers-Squibb. El Dr. Santos Castañeda ha recibido ayudas de educación e investigación de los laboratorios Abbott, MSD y Pfizer.


