




Hemophagocytic syndrome (HS) occurs in autoimmune diseases and belongs to the hemophagocytic lymphohistiocytosis group of diseases. This paper describes the features of 2 patients with systemic lupus erythematosus (SLE) who presented HS as the initial clinical manifestation.
Clinical observationsBoth patients had prolonged fever not associated to an infectious process and did not respond to broad-spectrum antibiotics.
DiscussionThe diagnosis of HS secondary to SLE is complicated, because it has some features in common, but HS is characterized by hyperferritinemia, hipofibrinogemia, hypertriglyceridemia and a decrease in the erythrocyte sedimentation rate, unlike SLE. HS treatment when associated to SLE is not well established, but steroids and/or immunoglobulins are effective as the initial treatment, and in refractory cases, cyclosporine or cyclophosphamide may be associated.
ConclusionsHS can be the initial manifestation of SLE and should be suspected in patients with organ enlargement, cytopenias, clotting disorders, liver disorders and prolonged fever unresponsive to antibiotics. Anakinra may be a treatment option in adult HS associated to SLE.
El síndrome hemofagocítico (SH) se produce en enfermedades autoinmunes y pertenece al grupo de enfermedades linfohistiocitosis hemofagocítica. El presente trabajo describe las características de 2 pacientes con lupus eritematoso sistemico (LES) que comenzaron con SH.
Observaciones clínicasAmbos pacientes presentaron fiebre prolongada no asociada a proceso infeccioso y que no respondía a antibióticos.
DiscusiónEl diagnóstico de SH secundario a LES es complicado, ya que presentan características comunes; sin embargo, el SH presenta hiperferritinemia, hipofibrinogenemia, hipertrigliceridemia y descenso de la velocidad de sedimentación globular a diferencia del LES. El tratamiento no está bien establecido, pero los corticoides y/o inmunoglobulinas son efectivos en el tratamiento inicial, y en casos refractarios la ciclosporina o la ciclofosfamida se pueden asociar.
ConclusionesEl SH puede ser la manifestación inicial del LES y debe sospecharse en pacientes con organomegalias, citopenias, trastornos en la coagulación, alteraciones hepáticas y fiebre prolongada que no responde a antibióticos. Anakinra puede ser una opción de tratamiento en el SH secundario al LES adulto.
Hemophagocytic syndrome (HS) occurs in autoimmune diseases, and belongs to the large group of lymphohystiocytocis hemophagocytic diseases (LHH)1Table 1 shows the classification of LHH. The familial form is autosomal recessive, so family history is often negative. The estimated incidence is 1 in 50000 live births.2,3 The secondary form is the result of an intense and uncontrollable immune reaction caused by infection, exposure to drugs or other causes.3
Classification of LHH.
| Genetic or primary LHH |
| Familiar LHH |
| Known genetic defects: for example, perforin or other |
| unknown genetic defects |
| Immune deficiencies |
| Chediak-Higashi |
| Griscelli Syndrome |
| X-linked lymphoproliferative syndrome |
| Acquired or secondary LHH |
| Secondary to infections |
| Secondary to endogenous products |
| secondary to rheumatic diseases |
| secondary to neoplasms |
LHH is characterized by the proliferation and activation of T cells and macrophages, which produce an excessive inflammatory response and hypersecretion of cytokines such as interferon-gamma, tumor necrosis factor, interleukin 1 (IL-1), IL-6, IL-10, IL-12, IL-18, and macrophage colony stimulating factor.1,4 The etiology is unknown, although some drugs and viruses have been postulated as possible triggers, such as the Epstein–Barr virus.4 Furthermore, this disease can occur spontaneously or as a complication of the underlying disease, or it may be caused by infection or a change in treatment.5
Clinically, patients present with prolonged fever that is unresponsive to broad-spectrum antibiotics, progressive pancytopenia, enlarged liver or spleen, liver dysfunction, coagulopathy and hyperferritinemia.1,4 The difference between HS and the rest of the LHH is marked hyperferritinemia, cytopenia at the onset, which is usually mild, and more pronounced coagulopathy.2,3 The pathognomonic feature is the presence of macrophages phagocytosing hematopoietic cells in bone marrow (BM), spleen and/or lymph.4Table 2 shows the diagnostic criteria of LHH.1
Criteria for the Diagnosis of LHH.
| Fever (temperature greater than 38.5°C) for 7 days or more |
| Spleen enlargement |
| Cytopenia in 2 or more cell lines |
| Hemoglobin <9g/dl |
| Platelets <100000/μL |
| Neutrophils <1000/μL |
| Hypertriglyceridemia (>160mg/dl) or hypofibrinogenemia (<150mg/dl) |
| Ferritin ≥500μg/l |
| Hepatitis |
| Soluble CD 25 (sIL-2 receptor) >2400IU/ml |
| Decreased or absent NK cell activity |
| Hemophagocytic cells in bone marrow, spleen or lymph nodes |
At least 5 criteria should be met for LHH diagnosis.
HS can occur at any stage of life. In the pediatric population it is more common in systemic lupus erythematosus (SLE) and juvenile idiopathic arthritis (JIA), especially the systemic variety. In the adult population it may appear associated to adult onset Still's disease, during the course of SLE and5 in several vasculitic syndromes. Prevalence of HS secondary to SLE is estimated between 0.9% and 4.6%, although these figures are underestimated.6,7
This paper describes the clinical, laboratory, treatment and progression data of 2 patients with SLE who presented HS, admitted to the University Hospital of Donostia, Spain.
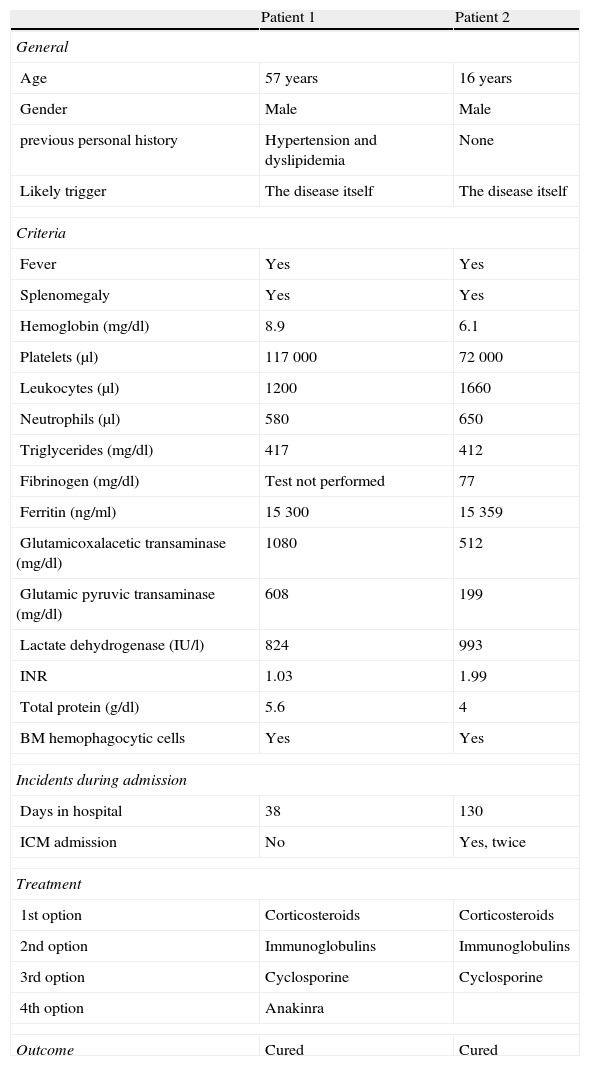
Clinical CasesTable 3 is a summary of the most important data found in both patients.
Demographic, Clinical, Laboratory and Treatment Data of Both Patients.
| Patient 1 | Patient 2 | |
| General | ||
| Age | 57 years | 16 years |
| Gender | Male | Male |
| previous personal history | Hypertension and dyslipidemia | None |
| Likely trigger | The disease itself | The disease itself |
| Criteria | ||
| Fever | Yes | Yes |
| Splenomegaly | Yes | Yes |
| Hemoglobin (mg/dl) | 8.9 | 6.1 |
| Platelets (μl) | 117000 | 72000 |
| Leukocytes (μl) | 1200 | 1660 |
| Neutrophils (μl) | 580 | 650 |
| Triglycerides (mg/dl) | 417 | 412 |
| Fibrinogen (mg/dl) | Test not performed | 77 |
| Ferritin (ng/ml) | 15300 | 15359 |
| Glutamicoxalacetic transaminase (mg/dl) | 1080 | 512 |
| Glutamic pyruvic transaminase (mg/dl) | 608 | 199 |
| Lactate dehydrogenase (IU/l) | 824 | 993 |
| INR | 1.03 | 1.99 |
| Total protein (g/dl) | 5.6 | 4 |
| BM hemophagocytic cells | Yes | Yes |
| Incidents during admission | ||
| Days in hospital | 38 | 130 |
| ICM admission | No | Yes, twice |
| Treatment | ||
| 1st option | Corticosteroids | Corticosteroids |
| 2nd option | Immunoglobulins | Immunoglobulins |
| 3rd option | Cyclosporine | Cyclosporine |
| 4th option | Anakinra | |
| Outcome | Cured | Cured |
57-year-old male patient with no history of importance, beginning two months prior to admission with general malaise, myalgia, pleuritic left chest pain and coughing with green phlegm; he went to the family physician who initially treated him with amoxicillin and ibuprofen and saw some improvement. The patient continued presenting the symptoms as well as fever without chills and constitutional syndrome, losing 10kg, so he decided to visit a private clinic (PC).
While in the PC, antibiotics (amoxicillin/clavulanic acid and levofloxacin) were started, and an associated infectious process ruled out (blood, urine, cerebrospinal fluid cultures and serology were negative for virus and bacteria). He progressed unfavorably with persistent fever, lymphadenopathy and splenomegaly with laboratory tests showing pancytopenia, hyperferritinemia, elevated liver enzymes and renal failure, all progressive, so for these reasons he was transferred to our hospitals hematology department.
At our hospital treatment with antibiotics was continued but no remission of fever was seen, with progressive worsening of pancytopenia, renal failure and hepatitis; hypertriglyceridemia was also added. Biopsy of a lymph node showed no signs of malignancy and a BM biopsy evidenced only showed macrophages was performed. LHH of uncertain etiology was diagnosed. Treatment was initiated with immunoglobulin and high-dose corticosteroids with improvement seen at the beginning. Subsequently the patient presented antinuclear antibody 1/640 with a homogeneous pattern and high titer anti-DNA, so the patient was sent to rheumatology.
After assessment by rheumatology and reviewing the patients medical history, we documented urine sediment with hematuria, pyuria, proteinuria and granular casts, hypocomplementemia, a direct Coombs test without evidence of hemolysis and a pericardial effusion seen on an echocardiogram performed at the PC. The diagnosis of HS secondary to SLE was reached and the patient was transferred to the department of rheumatology.
During admission in rheumatology, intermittent arthritis, facial erythema, oral ulcers, proteinuria 2.4g/day and persistent pancytopenia was seen, so cyclosporine 3mg/kg/day was added, with improvement. Renal biopsy was performed before discharge, showing glomerulonephritis stage iii.
Clinical Case 216-year-old male patient who began two months prior to admission with a constitutional syndrome with loss of 13kg, night sweats, afternoon fever, severe asthenia and rash on the chest, seen by the Infectious Diseases Unit, receiving empirical treatment with tetracycline without improvement.
On admission, the patient was feverish and had cervical lymphadenopathy. Initially tuberculosis was ruled out and the possibility of a lymphoproliferative disorder considered, so a BM and a lymph node biopsy were performed with negative results. The patient presented positive serology for Rickettsia which was interpreted as a false positive, and positive serology for Leishmania (IgG 1/256); amphotericin B was installed for 5 days at 3mg/kg/day, with improvement at the beginning, but leading to elevated pancreatic enzymes, therefore obliging its suspension due to secondary pancreatitis. Furthermore, during admission, ANA >1/1280, anti-DNA >600, positive anti-ENA, anti-Scl70 and anti-histones were seen, so an evaluation by Rheumatology was sought. Treatment was initiated with corticosteroids 1mg/kg/day for suspected connective tissue disease, with initial improvement and remission of fever.
In spite of amphotericin B suspension and continuing with corticosteroids, the patient presented abdominal pain, vomiting, worsening of pancreatic enzymes levels, fever and a generalized seizure. We performed a cranial CT with contrast which was normal and an abdominal CT with contrast, showing splenic infarcts, hepatomegaly, increased abdominal lymphadenopathy and free peritoneal fluid in the Douglas recess, so we decided to transfer the patient to Intensive Care Medicine (ICM). While in the Infectious Diseases Unit the patient presented anemia, leukopenia, lymphopenia, acute hepatitis and hypocomplementemia, the BM biopsy reported a slightly hypercellular marrow without evidence of infiltration and increased macrophages with some phagocytosis but not regarded as hemophagocytic; he was also treated with antibiotic, ceftriaxone and piperacillin–tazobactam.
While in ICM we began cyclosporine 3mg/kg/day for suspected SLE or SHSH, with improvement at the beginning. The general examination on admission revealed generalized muscle atrophy, mobile painless lymphadenopathy, oral ulcers, swollen abdomen but no resistance, macules on the trunk and livedo reticularis on the skin of the lower extremities, as well as anuria. The laboratory studies showed worsening of the data described, with further hyperferritinemia and elevated LDH. Blood cultures, stool cultures, urine and cerebrospinal fluid cultures were negative and he was serologically negative for viruses and bacteria, presenting negative DNA for Leishmania.
HS secondary to SLE complicated with multiple organ dysfunction syndrome was diagnosed. Treatment with antibiotics, corticosteroids, cyclosporine, and immunoglobulins was continued and anakinra was added. The outcome was favorable, with complete neurological recovery and progressive decrease of the analytical parameters. Later, during his stay in ICM he presented sinus tachycardia and bradyarrhythmias attributed to cardiotoxicity due to cyclosporine, which was suspended.
Regarding HS, he progressed well, but then presented specific bacterial peritonitis which required antibiotics and suspension of anakinra. The patient was discharged from ICM and sent home due to improvement, but was readmitted within 7 days for worsening of the infection, requiring four surgical peritoneal lavages, discharged from General Surgery and subsequently discharged from the hospital.
Both patients had prolonged fever unresponsive to broad-spectrum antibiotics. During admission, any associated infectious process was discarded. Both patients met 7 of the 9 criteria for LHH. The 2 tests that were not met were quantifying natural killer cells and sIL-2 receptor, tests that are not performed at our center.
DiscussionThe diagnosis of HS secondary to SLE is complicated because they share some common characteristics such as fever, pancytopenia, lymphadenopathy, neurological, joint, skin, kidney and cardiac manifestations 8. Table 4 shows the preliminary diagnostic criteria for macrophage activation syndrome as a complication of juvenile SLE proposed by Parodi et al.5 These have a sensitivity and specificity of 92.1% and 90.9% respectively, and an OR of 116.7% with a confidence interval between 21.9 and 621.6 at 95% confidence.5 These criteria have advantages and disadvantages regarding the criteria proposed for LHH. Advantages are the fact that a BM biopsy should be performed only in cases of diagnostic doubt, which seems reasonable because it is very invasive and often delays diagnosis and initiation of treatment. The most important disadvantage is that these criteria are not able to distinguish from an infectious complication.
Preliminary Diagnostic Criteria for Macrophage Activation Syndrome Complicating SLE.
| Clinical criteria |
| Fever (>38°C) |
| Hepatomegaly (≥3cm below the rib margin) |
| Splenomegaly (≥3cm below the rib margin) |
| Hemorrhagic manifestations (purpura, easy bruising or bleeding gums) |
| Dysfunction of the central nervous system (irritability, disorientation, lethargy, headache, convulsions or coma) |
| Laboratory criteria |
| Cytopenia in 2 or more cell lines (4 leukocytes×109/l, hemoglobin 90g/l or platelets 150×109/l) |
| Increased aspartate aminotransferase (>40units/l) |
| Increased lactate dehydrogenase (>567units/l) |
| Hypofibrinogenemia (fibrinogen <1.5g/l) |
| Hypertriglyceridemia (triglycerides >178mg/dl) |
| Hyperferritinemia (ferritin >500μg/l) |
| Histopathological criteria |
| Evidence of hemophagocytic macrophages in bone marrow |
For diagnosis, the simultaneous presence of at least one clinical criterion and at least 2 laboratory criteria is required. The bone marrow biopsy may be needed only in doubtful cases.
Certain laboratory parameters help us discern between HS and active SLE. Thrombocytopenia is a better indicator HS than leukopenia or anemia. Hyperferritinemia seems to have the strongest ability to discriminate between these two possibilities with a sensitivity and specificity of almost 100%.5,9
HS is more frequent in pediatric patients with rheumatic diseases, especially with JIA. However, in recent years it is ever more common in SLE, probably because before it was underdiagnosed.5,7,10 HS in adult SLE is associated with the disease onset in most patients, as in the case of our patients; this is an important difference with systemic JIA, in which it appears in more advanced stages of the disease.11,12
The goal of treatment is to stop the excessive inflammatory response. Treatment HS secondary to SLE is not well established, as it is in JIA.8,12 Initial treatment is parenteral corticosteroids. Intravenous immunoglobulins are an option, especially when an associated viral infection1 is suspected. Early introduction of cyclophosphamide is also an option, since corticosteroids appear to be insufficient and since it is a drug used in severe SLE.11,13 In corticosteroid refractory cases, oral or parenteral cyclosporine may be associated, and this has been effective in patients with systemic JIA and juvenile SLE.1,14 Cyclophosphamide and cyclosporine have shown no significant differences in treatment.5 Biological treatments are used when the inflammatory process is not adequately controlled with steroids and/or immunosuppressive drugs. Among biological treatment options infliximab could be used, which has proven effective in patients with SLE associated HS.15 Another option could be rituximab, which has proven effective in patients with severe5 pancytopenia. In one of our patients we used anakinra, a treatment has proven effective in HS secondary to systemic JIA; it is also a drug used daily, so it could be suspended at any time for any infectious complication.4,16
Other measures to consider during immunosuppressive therapy are: gastroprotection, cotrimoxazole prophylaxis, oral antifungal drugs and evaluation of the use of antiretroviral drugs.1In cases of reactivation, broad-spectrum antibiotics should be started, although some authors mention that treatment should start with immunosuppression.1,17
ConclusionsHS can be the initial clinical manifestation of SLE and should be suspected in patients with enlarged liver or spleen, cytopenias, clotting disorders, liver disorders and prolonged fever unresponsive to broad-spectrum antibiotics.
Anakinra may be a treatment option in adult HS secondary to SLE refractory to corticosteroids, immunoglobulins, and cyclosporine.
Ethical ResponsibilitiesProtection of persons and animalsThe authors state that no experiments were performed on persons or animals for this study.
Data confidentialityThe authors state that they have followed their workplace protocols regarding the publication of patient data and all patients included in the study have received enough information and have given their written informed consent to participate in the study.
Right to privacy and informed consentThe authors state that they have obtained informed consent from patients and/or subjects referred to in this article. This document is in the possession of the corresponding author.
Conflict of InterestThe authors declare no conflicts of interest.
Please cite this article as: Egües Dubuc CA, Uriarte Ecenarro M, Meneses Villalba C, Aldasoro Cáceres V, Hernando Rubio I, Belzunegui Otano J. Síndrome hemofagocítico como manifestación clínica inicial del lupus eritematoso sistémico. Reumatol Clin. 2013;10:321–324.


