



Muckle-Wells syndrome is a systemic autoinflammatory disease included in the group of hereditary periodic febrile syndromes.
We report the case of a patient with this rare disease to call the attention to the singularity of this condition, its low incidence, its atypical presentation and the subsequent delay in the diagnosis, which is reached when late and devastating consequences have taken place.
In this case, the first-line therapy, anti-interleukin 1 (IL-1), failed to control the disease. Nevertheless, the IL-6 inhibitor, tocilizumab, proved effective, achieving the total remission of nephrotic syndrome associated with AA secondary amyloidosis, changing the bleak prognosis of this disease.
El síndrome de Muckle-Wells es una enfermedad autoinflamatoria sistémica incluida dentro del grupo de fiebres periódicas hereditarias.
Presentamos a un paciente con esta enfermedad para llamar la atención sobre la singularidad de esta entidad, su baja frecuencia y presentación atípica, que conllevan generalmente demora en el diagnóstico, cuando ya hay consecuencias tardías y muchas veces devastadoras.
En este caso, la terapia de primera línea antiinterleucina 1 (IL-1) no consiguió frenar la enfermedad, consiguiendo, sin embargo, su control el inhibidor de IL-6 tocilizumab, mostrándose eficaz en la remisión total del síndrome nefrótico asociado a amiloidosis secundaria AA, cambiando su oscuro pronóstico.
Muckle-Wells syndrome is a rare hereditary disease with an autosomal dominant transmission pattern. It is caused by the mutation of the NLRP3 gene (also referred to as CIAS1), which encodes cryopyrin, a protein responsible for the regulation of the production of inflammatory cytokines, mainly interleukin (IL)-1β, which leads to a persistent and uncontrollable systemic inflammation.1 There are 3 types of cryopyrin-associated periodic syndromes (CAPS) of increasing severity: familial cold autoinflammatory syndrome, Muckle-wells syndrome and chronic infantile neurologic, cutaneous, articular (CINCA) syndrome and neonatal onset multisystem inflammatory disease (NOMID).
There are no clearly defined diagnostic criteria but, according to the medical literature, its diagnosis is based on 3 findings. In the first place, the clinical onset begins with episodes characterized by fever, rash and/or urticaria and arthralgia and/or arthritis associated with abdominal pain and previous conjunctivitis and/or uveitis. These episodes are self-limiting and recurrent, and have a duration of 2–5 days. Subsequently, the patient may be found to have progressive sensorineural deafness and/or secondary (AA) amyloidosis (fundamentally with renal involvement); the first develops in approximately 60% of the patients, whereas the second occurs in around 25%.2
Treatment is initially based on nonsteroidal anti-inflammatory drugs (NSAID), colchicine and corticosteroids, in addition to anti-IL-1 therapies3 (mainly anakinra, rilonacept and canakinumab).4
Case ReportThe patient was a 25-year-old man who had had recurrent episodes of prolonged fever since he was 2 years old, occurring 1 to 2 times a year. The presence of hepatosplenomegaly had been detected in a pediatric examination. He also had joint and muscle pain in the absence of inflammatory signs; he had a few episodes of abdominal pain and evanescent rash on 2 occasions. There was no family history of recurrent fevers or connective tissue diseases. He was evaluated in the pediatrics, rheumatology, hematology and internal medicine departments, with an initial diagnosis of systemic onset Still's disease, which was treated with NSAID and paracetamol.
His laboratory tests showed persistent leukocytosis and elevation of acute phase reactants, C-reactive protein and erythrocyte sedimentation rate. However, the levels of ferritin, rheumatoid factor, antinuclear antibodies (ANA), the complement system and immunoglobulins were always negative or within the normal range.
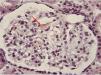
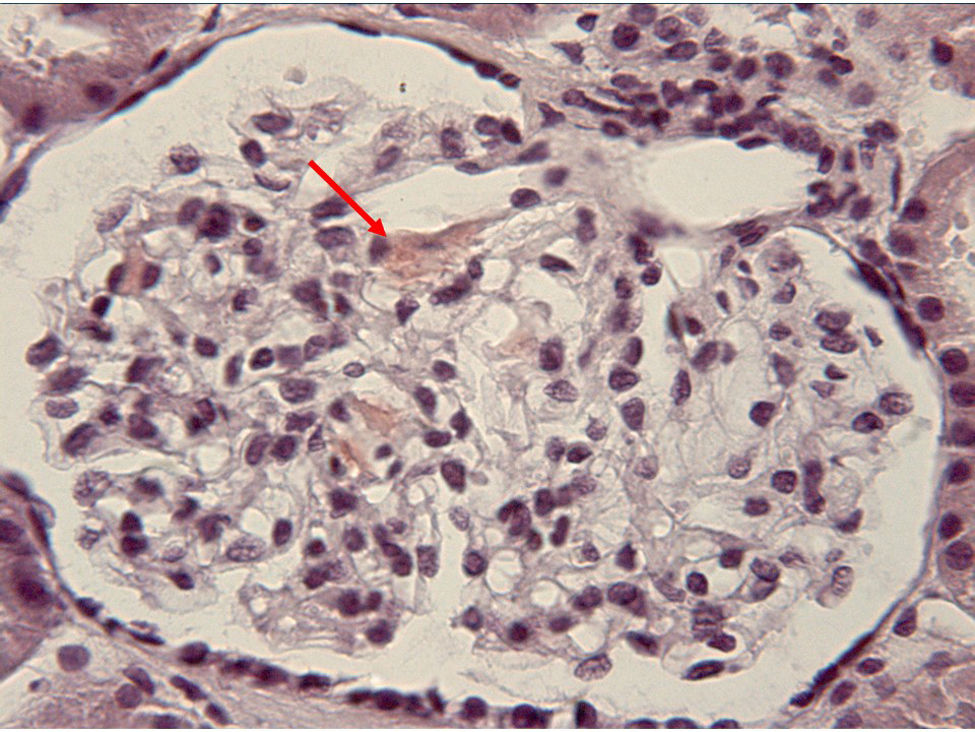
When the patient was 23 years old, the presence of proteinuria in the nephrotic range was detected, with no changes in the sediment. Given the suspicion of proteinuria in relation to the administration of NSAID or his underlying inflammatory process, we requested a renal biopsy, which revealed secondary amyloidosis (Fig. 1). At that time, with the above description of the clinical findings and analytical results, and after the development of renal amyloidosis, we decided to reconsider the diagnosis. We thought of the possibility that it could be a hereditary systemic autoinflammatory disease rather than Still's disease, and we requested genetic studies in that respect. As the result was positive for mutation of the NLRP3 gene, the diagnosis was Muckle-Wells syndrome.
Treatment was begun with subcutaneous anakinra at a dose of 100mg daily,4 but that failed to achieve adequate control of proteinuria. Given the poor response and following the finding of elevated IL-6 levels, anakinra was discontinued and treatment was initiated with tocilizumab (an IL-6 inhibitor). Over the past 6 years, the patient has been asymptomatic, with good control of the inflammation and normal renal function without proteinuria.
DiscussionSince infancy, our patient had had recurrent episodes of fever and arthralgia, in addition to episodes of abdominal pain and skin rash. Rheumatoid factor and ANA were negative, all of which is compatible both with Still's disease5 and cryopyrinopathies. The coexistence of hepatosplenomegaly, according to the criteria of Yamaguchi,6 indicated the diagnosis of Still's disease. It was not until the development of renal amyloidosis, 20 years later, that the diagnosis was reconsidered. The genetic study confirmed the diagnosis of Muckle-Wells syndrome.
ConclusionGiven the rarity of these syndromes in routine clinical practice, it is necessary to stress the importance of being aware of their different and sometimes incomplete forms of presentation and maintain a high index of suspicion. This will help in the design of an adequate differential diagnosis, in deciding the genetic tests that confirm the definitive diagnosis and in implementing a suitable biological therapy. This will help to curb, or at least slow, the advance of the disease and with it the development of complications – renal amyloidosis – that can effect the prognosis, as occurred in this case. Nevertheless, the IL-6 inhibitor, tocilizumab, proved effective in achieving the total remission of nephrotic syndrome, changing the prognosis of this disease.
Ethical DisclosuresProtection of human and animal subjectsThe authors declare that no experiments were performed on humans or animals for this study.
Confidentiality of dataThe authors declare that they have followed the protocols of their work center on the publication of patient data.
Right to privacy and informed consentThe authors declare that no patient data appear in this article.
Conflicts of InterestThe authors declare they have no conflicts of interest.
Please cite this article as: Solís Marquínez MN, García Fernández E, Morís de la Tassa J. Fiebres periódicas: de la enfermedad de Still al síndrome de Muckle-Wells. Reumatol Clin. 2019;15:e39–e40.


