


Many environmental factors have been associated with an increased risk of developing Rheumatoid Arthritis (RA), but so far smoking is the only environmental risk factor that has been extensively studied and widely accepted. Smoking is associated with an increased risk of developing seropositive RA (RF and/or ACPA). Recent studies show that tobacco smoking can influence disease phenotype, with the development of more aggressive disease and greater joint damage; but other studies show contradictory results. Recent data suggests that response to antirheumatic therapy in RA is worse in smokers. In this article we review different environmental factors that have been associated with an increased risk of developing RA, with a special interest in tobacco smoking.
Existen distintos factores ambientales implicados en la patogenia de la artritis reumatoide, aunque es el tabaco el factor más ampliamente estudiado y reconocido. El tabaquismo está asociado a un incremento del riesgo de artritis reumatoide seropositiva (FR y/o ACPA). Además estudios recientes ponen de manifiesto que el consumo de tabaco puede influir en la expresión clínica de la enfermedad, determinar un curso evolutivo más grave y una mayor destrucción articular, aunque no todos los estudios son concordantes. Datos recientes sugieren que la respuesta al tratamiento antirreumático sería peor en los enfermos fumadores. En el presente artículo se revisan los distintos factores ambientales que han sido implicados en la AR, con especial énfasis en el tabaquismo.
Rheumatoid arthritis (RA) is the most common chronic inflammatory joint disease, affecting about 0.5%–1% of the general population and causing progressive joint destruction, disability and reduced life expectancy. The etiology of RA is unknown and its pathogenesis is only partially explained to date. In recent years we have studied and identified multiple risk factors for its development. We know that genetic and environmental factors are involved and their interaction may be decisive in the development of the disease. Among the environmental factors is tobacco, which has been extensively studied and is now recognized as an important risk factor for RA.1
Rheumatoid Arthritis and Genetic Risk FactorsFor over 30 years we have known that there is a genetic susceptibility factor in RA2 which contributes 50%–60% to the development of the disease.3 Several studies have reported the presence of the disease in between 2% and 4% in dizygotic twins and between 12% and 15% in monozygotic twins.3,4 Thus, those with a first degree relative with RA may have from 2 to 10 times greater risk of developing the disease than the general population.5 Initially, an association of RA with HLADRB1 *04 and some of its alleles (*0401, *0404, *0405 or *0408) was found, and later confirmed with other alleles HLADRB as *0101, *0102 or *010. Today we know that all these alleles encode the same amino acid sequence of the third hypervariable region of the beta chain of the HLA molecule, a region that is central to the process known as antigen recognition, and has been called rheumatoid epitope (RE) or shared epitope (SE). The presence of these alleles not only increases the risk of developing RA, especially seropositive disease (positive rheumatoid factor (RF) and/or antibodies against citrullinated proteins/peptides (ACPA)) but their presence in numerous studies has been associated to environmental factors such as tobbaco,6–10 as well as a worse prognosis, with a greater degree of joint destruction and presence of extra-articular11–13 manifestations. This poor prognosis is particularly evident when the RE is included within the HLA-DRB1 *04, especially with allele *0401. But HLA-DRB1 accounts for approximately one third of the genetic component of RA,14,15 and an intense search has been undertaken for other non-HLA genes associated with RA. In association studies of candidate genes many genetic polymorphisms that also contribute to the development of RA, but to a lesser extent thn the RE, have been identified. Between them, PTPN22 (a functional variant of the protein tyrosine phosphatase intracellular N22) stands out as the second largest susceptibility gene for the development of the RA, which doubles the risk of seropositive RA in heterozygous and four times in homozigotes.16,17 Other RA susceptibility genes in populations of European descent are STAT4,18,19 a key transcription factor in regulating the immune response involved in signaling pathways that promote differentiation of CD4 Th1 and Th,17 which are involved in the pathogenesis of RA and TRAF1/C5.20,21 The recent availability of whole genome studies (GWASs) has allowed the identification of more than 20 susceptibility locus for RA, whose contribution to genetic variation as a whole could be around 5%.20
Rheumatoid Arthritis and Non-genetic Risk FactorsAs mentioned, genetic factors warrant about 50% of the risk of RA, leaving the rest to other factors. We have studied various environmental factors, although the scientific evidence on their exact involvement is inconclusive in many cases. We review briefly the most important environmental factors.
Hormones. The higher prevalence of RA in women, especially during childbearing years and frequent disease improvement during pregnancy22 has led to the identification of the possible hormonal role in disease susceptibility. There is considerable controversy as to whether oral contraceptives decrease the risk of developing RA, with some studies finding a clear association,23–25 others,26–30 including a metanalysis31 not showing a lower incidence of RA in women treated with oral contraceptives. One such study noted, however, that early menarche or extended breastfeeding (more than 12 months total) decrease the risk of RA,29 although this last point also is controversial because some studies have observed the opposite effect.24 A recently published study indicates that early menopause favors the risk of developing RA.32 Moreover, in men with RA, male sex hormone levels, especially testosterone, have been found decreased, while no differences were observed in the levels of sex hormones in women with RA and healthy controls.33
Socioeconomic factors. Socioeconomic status influences the course of the disease, but may also determine an increased risk of developing it.34 There was an inverse association between level of formal education and socioeconomic status defined by work activity and the risk of RA.35,36
Dietary factors. It has been suggested that a Mediterranean diet, rich in fish, olive oil, cooked vegetables and fruit has a protective effect against RA which could be due to the high content of omega 3 in such a diet.37,38 Consumption of red meat would have no effect on the risk of developing RA.39
Vitamin D. Vitamin D has been widely studied regarding its involvement in various autoimmune diseases. Its role in relation to risk of developing RA is “misleading”,40,41 although there seems to be an inverse association between consumption of vitamin D and development of RA,42 and between serum levels of vitamin D and disease progression, showing increased activity of the disease and more disability at lower levels of vitamin D.43–45
Alcohol. According to a recently published Danish study, alcohol consumption may have a protective effect on RA, in a dose dependent manner,27 observing that the magnitude of risk reduction would be greater in smokers and bearers of the SE.46
Coffee. Multiple studies have analyzed the effect of coffee consumption on RA but the results are discordant,47–49 although there could be an increased risk of RA in relation to consumption of high doses of coffee (more than 10 cups a day).27
Infections. Several infectious agents have been studied and implicated in the development of RA based on a higher frequency of positive viral serology or viral presence in synovial fluid of RA patients; however, their role as triggering agents of the disease is still controversial. Perhaps these agents may have some involvement in the development of the disease in the context of genetic predisposition and not in isolation, but together with other interacting risk factors. It should be noted the great interest in recent years Porphyromonas gingivalis has aroused as a potential stimulus for the development of RA. P. gingivalis is the major causative agent of periodontitis, a disease that is more frequent, about double, in subjects with RA than in the healthy population.50 It is the only bacterium known to express the enzyme peptidylarginine-deiminase (PAD), responsible for citrullination of proteins,51 and produces chronic inflammation (characterized by the presence of proinflammatory cytokines and TNF) and erosive destruction of periodontal bone.52 Like RA, periodontal disease has been associated with HLA-DRB04.53
Silica. Exposure to crystalline silica is a well defined risk factor for RA. Silica is present in the mining, construction, ceramics, glass, as well as in the agriculture sector and in electronics, and doubles the risk of RA in an analysis adjusting for exposure to tobacco.54
Tobacco. Tobacco is the most widely studied and recognized environmental risk factor for development of RA. Over 20 years ago Vassey et al.30 first suggested its involvement in RA, unexpectedly observing this effect in their study on the effect of oral contraceptives in RA. Since then this effect of tobacco has been reproduced and confirmed in multiple studies and case-control cohorts.1,55–58 These studies examined the effect of tobacco on RA as a risk factor for its development and its strong interaction with genetic factors and ACPA, as well as its effect on clinical, radiological and response to disease modifying treatments, leading to a better understanding of the disease. We discuss in detail the effect of tobacco in the various aspects of RA.
Rheumatoid Arthritis and TobaccoTobacco and ImmunityThe use of tobacco affects multiple organs including the respiratory and cardiovascular systems, but also affects the immune system by producing an inflammatory response. It has been observed that tobacco affects both the humoral and cellular immune response and could have both pro-and anti-inflammatory effects as immunosuppressants through diverse59 mechanisms. On one side it has been described that it may increase the inflammatory response observed in smokers by increasing serum fibrinogen, the activity of autoreactive B cells and an increase in acute phase reactants and pro-inflammatory cytokines such as TNF-alpha, IL6 as well as circulating polymorphonuclear cells. However it is also known for its immunosuppressive effects, such as a reduction of circulating immunoglobulins and inhibition of cytokines such as IL-1B, IL-2 and gamma-interferon or IL-8 release by endothelial cells. It would also have effects on dendritic cells and antigen presentation and inhibition of macrophage function against intracellular microorganisms.60
It has been observed that the use of tobacco modulates cell proliferation and death, inducing new epitopes either directly through the oxidation of existing epitopes or indirectly by interfering with the elimination of apoptotic cells with the consequent exposure of sequestered intracellular antigens by the immune system and stimulating the population of antigen presenting cells present in the lungs, thereby amplifying new antigen presenting capacity,61 facilitating the development of autoimmunity. Furthermore tobacco smoke contains very high concentrations of free radicals and may also increase the generation and activation of endogenous free radicals. These toxins interact with DNA and may cause mutation or gene activation that ultimately can trigger autoimmune phenomena and autoinmune disease.62 Thus, use of tobacco has been causally linked to the development and expression of multiple autoimmune diseases including RA,30,56 systemic lupus erythematosus,63,64 multiple sclerosis,65,66 Graves disease67,68 and primary biliary cirrhosis69 among others.
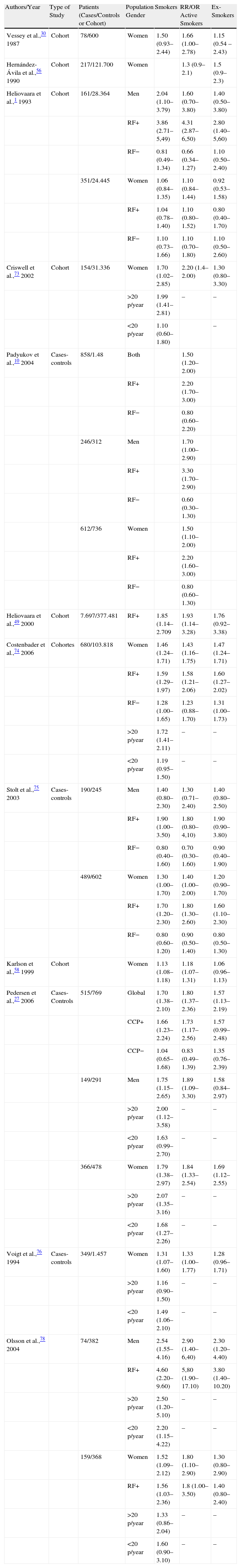
Tobacco and Risk of Rheumatoid ArthritisAs previously mentioned, after the first description of the association of tobacco with RA in 1987,30 two large prospective studies were then published in the 1990s, the first,56 a cohort study of 121700 nurses in which a slight increased risk of developing RA in relation to non-smokers was observed, with an RR for active smokers of 1.3 (95% CI 0.9–2.1) and RR for ex-smokers of 1.5 (95% CI 0.9–2.3), as well as a second study1 of more than 50000 subjects that showed an increased risk of developing seropositive RA (RF positive) in active smokers (RR 3.8, 95% CI 2.0–6.9) and former smokers (RR 2.6, 95% CI 1.3–5.3) compared with nonsmokers. Many other studies confirmed this association between tobacco and RA10,57,70–72 and several of them noted that the excess risk persisted after quitting smoking for 10 or even 20 years,27,49,73–78 that the probably years of consumption of tobacco weighed more than the number of cigarettes smoked daily in relation to the risk of RA58 and that a dose effect could be seen, i.e. the greater the number of cigarettes per day and pack/years smoked increased the risk of RA.27,74Table 1 summarizes the epidemiological studies on smoking and risk of RA (Sugiyama et al.79 modified).
Epidemiological Studies on the Risk of Rheumatoid Arthritis and Smoking.
| Authors/Year | Type of Study | Patients (Cases/Controls or Cohort) | Population Gender | Smokers | RR/OR Active Smokers | Ex-Smokers |
| Vessey et al.,30 1987 | Cohort | 78/600 | Women | 1.50 (0.93–2.44) | 1.66 (1.00–2.78) | 1.15 (0.54 –2.43) |
| Hernández-Ávila et al.,56 1990 | Cohort | 217/121.700 | Women | 1.3 (0.9–2.1) | 1.5 (0.9–2.3) | |
| Heliovaara et al.,1 1993 | Cohort | 161/28.364 | Men | 2.04 (1.10–3.79) | 1.60 (0.70–3.80) | 1.40 (0.50–3.80) |
| RF+ | 3.86 (2.71–5,49) | 4.31 (2.87–6,50) | 2.80 (1.40–5,60) | |||
| RF− | 0.81 (0.49–1.34) | 0.66 (0.34–1.27) | 1.10 (0.50–2.40) | |||
| 351/24.445 | Women | 1.06 (0.84–1.35) | 1.10 (0.84–1.44) | 0.92 (0.53–1.58) | ||
| RF+ | 1.04 (0.78–1.40) | 1.10 (0.80–1.52) | 0.80 (0.40–1.70) | |||
| RF− | 1.10 (0.73–1.66) | 1.10 (0.70–1.80) | 1.10 (0.50–2.60) | |||
| Criswell et al.,73 2002 | Cohort | 154/31.336 | Women | 1.70 (1.02–2.85) | 2.20 (1.4–2.00) | 1.30 (0.80–3.30) |
| >20 p/year | 1.99 (1.41–2.81) | – | – | |||
| <20 p/year | 1.10 (0.60–1.80) | – | ||||
| Padyukov et al.,10 2004 | Cases-controls | 858/1.48 | Both | 1.50 (1.20–2.00) | ||
| RF+ | 2.20 (1.70–3.00) | |||||
| RF− | 0.80 (0.60–2.20) | |||||
| 246/312 | Men | 1.70 (1.00–2.90) | ||||
| RF+ | 3.30 (1.70–2.90) | |||||
| RF− | 0.60 (0.30–1.30) | |||||
| 612/736 | Women | 1.50 (1.10–2.00) | ||||
| RF+ | 2.20 (1.60–3.00) | |||||
| RF− | 0.80 (0.60–1.30) | |||||
| Heliovaara et al.,49 2000 | Cohort | 7.697/377.481 | RF+ | 1.85 (1.14–2.709 | 1.93 (1.14–3.28) | 1.76 (0.92–3.38) |
| Costenbader et al.,74 2006 | Cohortes | 680/103.818 | Women | 1.46 (1.24–1.71) | 1.43 (1.16–1.75) | 1.47 (1.24–1.71) |
| RF+ | 1.59 (1.29–1.97) | 1.58 (1.21–2.06) | 1.60 (1.27–2.02) | |||
| RF− | 1.28 (1.00–1.65) | 1.23 (0.88–1.70) | 1.31 (1.00–1.73) | |||
| >20 p/year | 1.72 (1.41–2.11) | – | – | |||
| <20 p/year | 1.19 (0.95–1.50) | – | – | |||
| Stolt et al.,75 2003 | Cases-controls | 190/245 | Men | 1.40 (0.80–2.30) | 1.30 (0.71–2.40) | 1.40 (0.80–2.50) |
| RF+ | 1.90 (1.00–3.50) | 1.80 (0.80–4,10) | 1.90 (0.90–3.80) | |||
| RF− | 0.80 (0.40–1.60) | 0.70 (0.30–1.60) | 0.90 (0.40–1.90) | |||
| 489/602 | Women | 1.30 (1.00–1.70) | 1.40 (1.00–2.00) | 1.20 (0.90–1.70) | ||
| RF+ | 1.70 (1.20–2.30) | 1.80 (1.30–2.60) | 1.60 (1.10–2.30) | |||
| RF− | 0.80 (0.60–1.20) | 0.90 (0.50–1.40) | 0.80 (0.50–1.30) | |||
| Karlson et al.,58 1999 | Cohort | Women | 1.13 (1.08–1.18) | 1.18 (1.07–1.31) | 1.06 (0.96–1.13) | |
| Pedersen et al.,27 2006 | Cases-Controls | 515/769 | Global | 1.70 (1.38–2.10) | 1.80 (1.37–2.36) | 1.57 (1.13–2.19) |
| CCP+ | 1.66 (1.23–2.24) | 1.73 (1.17–2.56) | 1.57 (0.99–2.48) | |||
| CCP− | 1.04 (0.65–1.68) | 0.83 (0.49–1.39) | 1.35 (0.76–2.39) | |||
| 149/291 | Men | 1.75 (1.15–2.65) | 1.89 (1.09–3.30) | 1.58 (0.84–2.97) | ||
| >20 p/year | 2.00 (1.12–3.58) | – | – | |||
| <20 p/year | 1.63 (0.99–2.70) | – | – | |||
| 366/478 | Women | 1.79 (1.38–2.97) | 1.84 (1.33–2.54) | 1.69 (1.12–2.55) | ||
| >20 p/year | 2.07 (1.35–3.16) | – | – | |||
| <20 p/year | 1.68 (1.27–2.26) | – | – | |||
| Voigt et al.,76 1994 | Cases-controls | 349/1.457 | Women | 1.31 (1.07–1.60) | 1.33 (1.00–1.77) | 1.28 (0.96–1.71) |
| >20 p/year | 1.16 (0.90–1.50) | – | – | |||
| <20 p/year | 1.49 (1.06–2.10) | – | – | |||
| Olsson et al.,78 2004 | 74/382 | Men | 2.54 (1.55–4.16) | 2.90 (1.40–6,40) | 2.30 (1.20–4.40) | |
| RF+ | 4.60 (2.20–9.60) | 5,80 (1.90–17.10) | 3.80 (1.40–10.20) | |||
| >20 p/year | 2.50 (1.20–5.10) | – | – | |||
| <20 p/year | 2.20 (1.15–4.22) | – | – | |||
| 159/368 | Women | 1.52 (1.09–2.12) | 1.80 (1.10–2.90) | 1.30 (0.80–2.90) | ||
| RF+ | 1.56 (1.03–2.36) | 1.8 (1.00–3.50) | 1.40 (0.80–2.40) | |||
| >20 p/year | 1.33 (0.86–2.04) | – | – | |||
| <20 p/year | 1.60 (0.90–3.10) | – | – |
Modified from Sugiyama et al.79
Approximately one third of ACPA positive RA patients could be attributed to the consumption of tobacco. In a recent study, smoking would be responsible for 35% of ACPA positive RA (95% CI 25%–45%), this effect being greater in men than in women (42% and 31%, respectively) and an even greater in the presence of two copies of the rheumatoid epitope (55% CI 95%: 3%–67%).80 This effect is comparable in intensity to that of smoking in coronary artery disease.
Tobacco, Antibodies to Citrullinated Proteins/Peptides/Rheumatoid Factor and HLA-DRB1It has been recently noted that the use of tobacco is selectively associated with increased risk of seropositive RA (RF and/or ACPA positive). Independent studies have shown that smoking increases the risk of seropositive but not seronegative RA, both in Caucasian (northern Europe)27,75,81–86 as Latinamerican9 populations. However, a study that analyzed an African-American population with RA failed to show this association87 and in one of three large U.S. cohorts, this association was observed exclusively in heavy smokers.88
The first studies examining the relationship between tobacco and RA observed the existence of a higher frequency of RF positivity and higher RF titers in smokers compared to those of non-smokers..8275,89 Later studies also confirmed that ACPA90,91 are more frequent in smokers with RA. It is known that there is a high correlation between the presence of ACPAs and RF, but some authors have analyzed both antibodies in smokers with RA, and a relationship has been seen with ACPA but not with RF.92 In another study where a subanalysis evaluating patients discordant for ACPA and RF was conducted, there were similar proportions of smokers in both groups (ACPA+/FR− vs ACPA−/FR+),90 suggesting an association of tobacco with both autoantibodies.
The increased risk of seropositive RA in smokers is associated with the presence of rheumatoid epitope (HLA-DRB1), showing that there is significant gene–environment interaction between the alleles of the RE and tobacco. This fact is clearly seen in a Swedish cohort of RA10 which analyzed the interaction between RE and smoking. The authors found that while in patients with one and two copies of the RE the relative risk of developing seropositive RA was 2.4 (95% CI 1.4–4.2) and 4.2 (95% CI 2.1–8.3), respectively, compared to those without RE, in smokers the risk increased to 5.5 (95% CI 3.0–10.0) in the presence of a copy of the RE and 15.7 (95% CI 7.2–34.2) in the presence of two copies of the RE. Neither RE nor tobacco nor the combination of both factors were associated with an increased risk of seronegative RA. Later studies found similar results8,92–95 (Table 2).
Studies on Relative Risk of Developing RA in Relation to Tobacco Use and HLADRB Genotype (Rheumatoid Epitope).
| Author | Population | Sample Size | Result |
| Padyukov et al.,10 2004 | Sweden | 858/1.048Cases-controls | RF+/ET−/RE−/− RR: 1RF+/ET+/RE−/− RR: 2.4 (1.3–4.6)RF+/ET−/RE−/+ RR: 2.4 (1.4-4.2)RF+/ET+/RE−/+ RR: 5.5 (3.0–10.0)RF+/ET−/RE+/+ RR: 4.2 (2.1–8.3)RF+/ET+/RE+/+ RR: 15.7 (7.2–34.2)RF−/ET−/RE−/− RR: 1RF−/ET+/RE−/− RR: 0.6 (0.6–1.1)RF−/ET−/RE−/+ RR: 0.9 (0.6–1.5)RF−/ET+/RE−/+ RR: 0.9 (0.5–1.7)RF−/ET−/RE+/+ RR: 0.7 (0.4–1.6)RF−/ET+/RE+/+ RR: 1.2 (0.5–3.0) |
| Kallberg et al.,8 2007EIRA study | Sweden | 1.970/2.405Cases-controls | Anti-CCP+/ET−/RE −/− RR: 1Anti-CCP+/ET+/RE−/− RR: 1.3 (0.7–2.2)Anti-CCP+/ET−/RE−/+ RR: 2.7 (1.6–4.7)Anti-CCP+/ET+/RE−/+ RR: 5.4 (3.3–8.9)Anti-CCP+/ET−/RE+/+ RR: 5.1 (2.7–9.8)Anti-CCP+/ET+/RE+/+ RR: 23.6 (12.8–43.8)Anti-CCP−/ET−/RE−/− RR: 1Anti-CCP−/ET+/RE−/− RR: 0.7 (0.5–1.1)Anti-CCP−/ET−/RE−/+ RR: 1.0 (0.6–1.5)Anti-CCP−/ET+/RE−/+ RR: 1.0 (0.7–1.5)Anti-CCP−/ET−/RE+/+ RR: 0.9 (0.5–1.8)Anti-CCP−/ET+/RE+/+ RR: 1.1 (0.6–2.2) |
| Pedersen et al.,89 2007 | Denmark | 309/445Cases-controls | ET−/RE−/− RR: 1ET+/RE−/+ RR: 1.74 (0.76–3.98)ET−/RE−/+ RR: 3.31 (1.58–6.96)ET+/RE−/+ RR: 5.07 (2.48–10.4)ET−/RE+/+ RR: 17.4 (7.22–41.8)ET+/RE+/+ RR: 57.4 (22.0–150) |
| Van der Helm-Van Mil et al.,90 2007 | Holland | 142/279RA vs undifferentiated arthritis | RE−/Anti-CCP−/ET− RR: 1RE−/Anti-CCP−/ET+ RR: 1 (0.5–2.2)RE−/Anti-CCP+/ET− RR: 7.5 (2.1–28.5)RE−/Anti-CCP+/ET+ RR: 9.4 (2.4–39.8)RE+/Anti-CCP−/ET− RR: 1RE+/Anti-CCP−/ET+ RR: 0.5 (0.2–1.2)RE+/Anti-CCP+/ET− RR: 3.3 (1.3–8.3)RE+/Anti-CCP+/ET+ RR: 8.0 (3.1–21.1) |
RR: relative risk; RF: Rheumatoid Factor; ET: Exposure to tobacco; RE: Rheumatoid epitope.
We hypothesize that the exposure to tobacco and the local inflammatory reaction and cell necrosis produced by it stimulates protein citrullination in the lungs thus providing a substrate for the activation of an immune response. Following this hypothesis we have studied the presence of citrullinated peptides and proteins in the bronchoalveolar lavage (BAL) of smokers noting in the latter that there is a significant increase relative to non-smokers.90 Along the same lines, another study has observed an increased expression of PAD in bronchoalveolar cells of smokers.96 The presence of genetic susceptibility factors for the citrullination of proteins in the lungs could facilitate the development of a local autoimmune response with production of anti-citrullinated peptide antibodies. At a later moment, the presence of articular inflammatory processes and therefore citrullinated peptides97 could form immunocomplexes with the circulating anti-citrulline, eventually triggering an immune response with release of inflammatory mediators (TNF, ILs).98
It is known that anti-citrullinated peptides (ACPA) can be detected in the serum years before the onset of clinical manifestations of RA.99,100 According to the hypothesis above, the prevalence of ACPA in smokers may be higher than in the general population, but this fact has been scarcely studied. Our group observed a similar frequency of ACPA in the serum of heavy smokers without RA and a control population (1.8% vs 1.9%), although in those with COPD, the frequency was slightly higher (3.7%).101 These observations are similar to those observed by Wood et al.102 group.
Tobacco and Clinical Course of DiseaseTobacco not only increases the risk of seropositive RA but also might influence the phenotype or clinical expression of disease.
RA patients who smoke have an earlier onset of disease.83,85,86,103–105 Most studies agree that other baseline characteristics such as duration and disease activity or disability, are comparable to those of non-smoking patients,104–106 although some observed an increased in baseline activity of the disease in smokers.83,84 The impact of tobacco in the clinical course of disease is unclear since some studies report that smokers have a worse outcome with greater disability and disease activity83,85,86,107; others show a similar pattern of disease in smokers and non-smokers.84,103,105 On the other hand, some studies indicate a higher frequency of extra-articular manifestations in smoking RA patients compared to non-smokers,108 mainly higher frequency of rheumatoid nodules82,83,87,103 and lung involvement.109,110
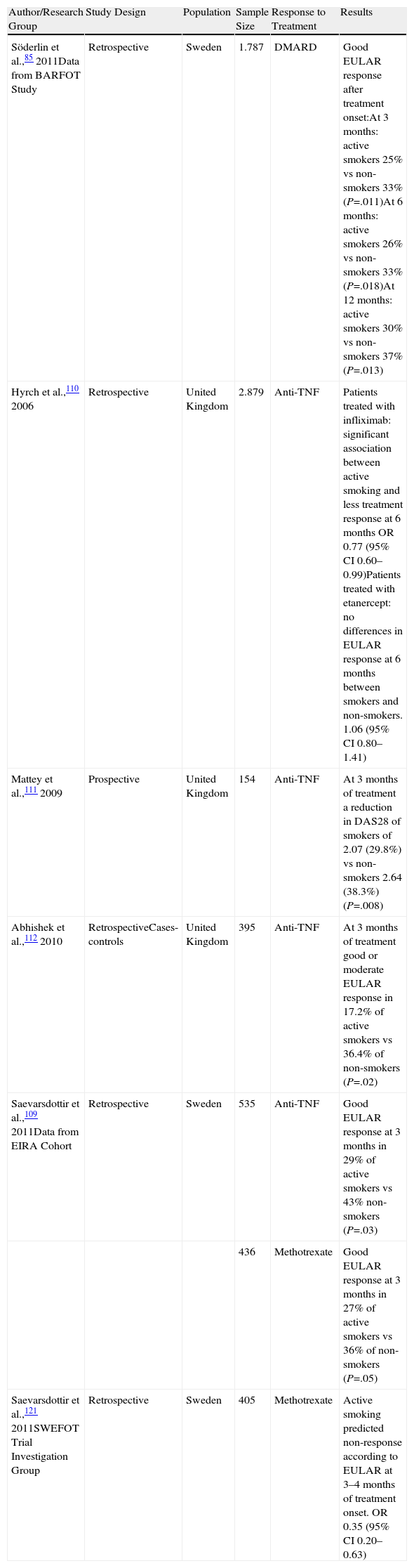
In recent years several studies have examined specifically whether smoking is a factor of poor response to antirheumatic treatment with DMARDs or TNF antagonists. A study shows that smokers used more DMARDs and higher doses than non-smokers,85 while two other studies confirmed that smoking is a factor of poor response to treatment with methotrexate in monotherapy.86,111 This would agree with data recently published from a Swedish registry study that evaluated the influence of tobacco in the response to methotrexate in a cohort of early onset RA; the authors found that after three months of treatment, nonsmokers achieved a significantly higher good EULAR response than smokers (36% vs 27%), with smokers less likely to achieve remission.112 This study also analyzed the response to anti-TNF as a first biologic after DMARD failure in smokers and nonsmokers and found that active smokers were less likely to achieve a good EULAR response at 3 months, succeeding only in 29% of current smokers compared with 43% of nonsmokers. In analyzing the probability of remission in both groups it found that active smokers had a tendency to a lower likelihood of achieving this objective at 3 months, this being significant at 6 months with an OR 0.54 (95% CI 0.30–0.99). Several other studies have analyzed the response to anti-TNF treatment in relation to consumption of tobacco and observed similar results, confirming a lower response to anti-TNF in smoking patients112–115 (Table 3). In addition, a subanalysis of the BeSt study identified several risk factors for reactivation of arthritis in patients who could discontinue infliximab after achieving good disease control, including having the RE, but also smoking.116
Effect of Tobacco on Treatment Response.
| Author/Research Group | Study Design | Population | Sample Size | Response to Treatment | Results |
| Söderlin et al.,85 2011Data from BARFOT Study | Retrospective | Sweden | 1.787 | DMARD | Good EULAR response after treatment onset:At 3 months: active smokers 25% vs non-smokers 33% (P=.011)At 6 months: active smokers 26% vs non-smokers 33% (P=.018)At 12 months: active smokers 30% vs non-smokers 37% (P=.013) |
| Hyrch et al.,110 2006 | Retrospective | United Kingdom | 2.879 | Anti-TNF | Patients treated with infliximab: significant association between active smoking and less treatment response at 6 months OR 0.77 (95% CI 0.60–0.99)Patients treated with etanercept: no differences in EULAR response at 6 months between smokers and non-smokers. 1.06 (95% CI 0.80–1.41) |
| Mattey et al.,111 2009 | Prospective | United Kingdom | 154 | Anti-TNF | At 3 months of treatment a reduction in DAS28 of smokers of 2.07 (29.8%) vs non-smokers 2.64 (38.3%) (P=.008) |
| Abhishek et al.,112 2010 | RetrospectiveCases-controls | United Kingdom | 395 | Anti-TNF | At 3 months of treatment good or moderate EULAR response in 17.2% of active smokers vs 36.4% of non-smokers (P=.02) |
| Saevarsdottir et al.,109 2011Data from EIRA Cohort | Retrospective | Sweden | 535 | Anti-TNF | Good EULAR response at 3 months in 29% of active smokers vs 43% non-smokers (P=.03) |
| 436 | Methotrexate | Good EULAR response at 3 months in 27% of active smokers vs 36% of non-smokers (P=.05) | |||
| Saevarsdottir et al.,121 2011SWEFOT Trial Investigation Group | Retrospective | Sweden | 405 | Methotrexate | Active smoking predicted non-response according to EULAR at 3–4 months of treatment onset. OR 0.35 (95% CI 0.20–0.63) |
OR: odds ratio; CI: confidence interval.
Tobacco consumption increases the metabolic rate and may be associated with resistance to anti-rheumatic treatment due to pharmacokinetic or pharmacodynamic interactions; for example it has been shown that smokers have lower levels of methotrexate polyglutamates, the active form of the drug, which correlates with clinical117 response. As for the response to anti-TNF differences in its pharmacokinetics or pharmacodynamics due to the use of tobacco have not been reported, although the presence of an increased production of TNF alpha by circulating T cells in smoking patients with RA has been observed, which could justify a lower response to treatment.118
Tobacco and Radiological ProgressionThe effect of smoking on radiographic progression of RA is not well established. Early studies addressing this aspect.81,8289 were a series of cross-sectional studies in cohorts of long-standing disease (mean 13 years or so) and found a significantly greater radiological progression in patients who smoked. Subsequent publications, both prospective and cross-studies, show conflicting results. In the study by Mattey et al.,119 which analyzes the effect of tobacco in a cohort of 164 patients with RA followed for 5 years, a significantly higher radiological damage Larsen score was seen in smokers compared with nonsmokers (83.1±47.2 and 104.7±49.9, respectively) as was a higher level of disability measured by the HAQ (1.39±0.8 vs 1.77±0.8, respectively). A Greek84 cohort of 287 patients with early onset RA obtained similar results, with greater radiological progression in active and former smokers than in nonsmokers after at least two years of monitoring. However, other prospective studies in large cohorts of recent onset RA do not corroborate these findings and radiological progression of the disease could be influenced by smoking.83,85,87,103,104,120 For example, in a multicenter study in 379 early onset RA patients,120 predictors of radiographic progression were explored in the univariate analysis and the major determinants of radiographic progression were the baseline Larsen score and serological anti-CCP and RF markers and, to a lesser extent, ESR, CRP, age, male gender and being a smoker (OR 1.6, 95% CI 1.0–2.5); however, in the multivariate analysis, smoking was not a independent risk factor, these being the baseline radiological damage, anti-CCP and ESR. Similarly Westhoff et al.85 in their cohort of 894 patients with recent onset RA found a weak association between smoking and radiographic progression, but this was not sustained in multivariate analysis.
However, in most of these studies, patients were not treated evenly, or prognostic factors of radiographic progression markers such as HLA-DRB genotype or the presence of ACPAs not analyzed. Recently our group analyzed, in a cohort of 156 RA patients followed for two years beginning and treated with DMARDs, whether smoking was a predictor of further radiological progression,105 and also analyzed multiple prognostic variables including clinical features, autoantibodies and HLADRB genotype. Multivariate analysis confirmed that active smoking (compared with the absence of that habit) was an independent factor of greater radiological progression assessed by the Larsen index with an OR of 4.3. Other factors for progression were female sex, the presence of HLA-DRB04 and radiographic damage at baseline.
ConclusionsWe have studied multiple RA predisposing environmental factors, although there is no doubt that tobacco is established as the most widely studied and with more scientific evidence on his involvement in this disease. Smoking could justify even a third of cases of seropositive RA, as the increased risk associated with tobacco, seems confined primarily to RA with positive autoantibodies (RF and/or ACPAs). The effect of tobacco on the activity of the disease and its clinical course is controversial, as on the progression of joint destruction, yet some studies indicate on a more severe phenotype of RA and greater radiological progression in smokers. Recent studies including large series of patients in registries, such as the Swedish or British, would confirm a worse response to treatment with DMARDs (methotrexate) and/or biological drugs (TNF antagonists) in smokers.
Despite this new evidence on the involvement of smoking in the predisposition and development of RA, there are still issues to resolve, such as the pathogenetic mechanisms involved, the effects of smoking cessation on the disease, the impact its abandonment could have in the general population in the future incidence of RA or its role in other forms of chronic arthritis.121–123 This does not question the fact that clinical rheumatologists must discourage smoking in all patients with RA. New knowledge on tobacco and RA and the fact that smoking is a known risk factor for atherosclerosis and other serious diseases, which are more prevalent in RA patients, fully warrants this advice.
Ethical disclosuresProtection of human and animal subjects. The authors declare that no experiments were performed on humans or animals for this investigation.
Confidentiality of Data. The authors declare that no patient data appears in this article.
Right to privacy and informed consent. The authors declare that no patient data appears in this article.
Conflict of interestThe authors declare no conflict of interest.
Please cite this article as: Ruiz-Esquide V, Sanmartí R. Tabaco y otros factores ambientales en la artritis reumatoide. Reumatol Clin. 2012;8:342–50.


