


No cabe duda de que los productos biológicos aportan un valor añadido a los sistemas de salud, aunque también plantean grandes interrogantes debido a su especial naturaleza, lo que obliga a ser muy rigurosos y exigentes en su control de calidad y seguimiento. Este hecho se ha visto reforzado por la entrada en escena de los fármacos biosimilares, cuyo menor coste está permitiéndoles alcanzar un mayor protagonismo en el mercado mundial. El propósito de este artículo es revisar en profundidad los principales interrogantes que se plantean en su producción, distribución y control, así como los aspectos más importantes relacionados con su seguridad en la práctica clínica. En este trabajo revisamos lo que representa la farmacovigilancia de estos productos, prestando especial atención a su trazabilidad, como herramienta fundamental para la detección precoz de acontecimientos adversos.
There is no doubt that biologic therapies provide added value for health systems. However, due to their special nature, they also raise some questions that make highly rigorous and demanding quality control and monitoring of their use indispensable. This circumstance is reinforced with the appearance on the scene of biosimilars, which, given their lower cost, are having an increasing impact on the international market. The purpose of this article is to review the major issues posed by their manufacture, distribution and control systems, as well as the most important aspects related to their safety in clinical practice. In this report, we assess the pharmacovigilance of these products, with special attention to traceability, as a key tool to enable earlier detection of adverse events.
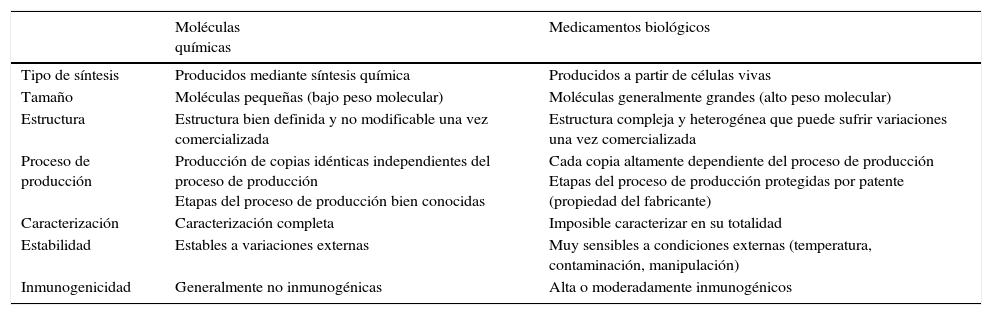
Los fármacos biológicos han supuesto un gran avance en el tratamiento de patologías complejas, como la artritis reumatoide (AR), psoriasis, enfermedad de Crohn, esclerosis múltiple, diabetes o el cáncer1-4, para las que, hasta su aparición, no disponíamos de opciones terapéuticas realmente eficaces5. Sin embargo, su alta complejidad en comparación con los fármacos de síntesis química tradicionales (tabla 1) obliga a tener una especial precaución en su manejo6. Por ello están incluidos en la lista de «medicamentos sujetos a seguimiento adicional» perteneciente al sistema de farmacovigilancia de la Unión Europea (UE)7. Además, habiendo llegado al momento de la expiración de las patentes de algunos de ellos y con la entrada en escena de los biosimilares, aparecen muchas interrogantes que trataremos de abordar en esta revisión.
Diferencias entre los fármacos de síntesis química y los medicamentos biológicos
| Moléculas químicas | Medicamentos biológicos | |
|---|---|---|
| Tipo de síntesis | Producidos mediante síntesis química | Producidos a partir de células vivas |
| Tamaño | Moléculas pequeñas (bajo peso molecular) | Moléculas generalmente grandes (alto peso molecular) |
| Estructura | Estructura bien definida y no modificable una vez comercializada | Estructura compleja y heterogénea que puede sufrir variaciones una vez comercializada |
| Proceso de producción | Producción de copias idénticas independientes del proceso de producción Etapas del proceso de producción bien conocidas | Cada copia altamente dependiente del proceso de producción Etapas del proceso de producción protegidas por patente (propiedad del fabricante) |
| Caracterización | Caracterización completa | Imposible caracterizar en su totalidad |
| Estabilidad | Estables a variaciones externas | Muy sensibles a condiciones externas (temperatura, contaminación, manipulación) |
| Inmunogenicidad | Generalmente no inmunogénicas | Alta o moderadamente inmunogénicos |
Los sistemas de farmacovigilancia tienen por objetivo la identificación, la cuantificación, la evaluación y la prevención de los riesgos del uso de los medicamentos una vez comercializados. Por lo tanto, están orientados a la toma de decisiones que permitan mantener en el mercado medicamentos con una relación beneficio-riesgo adecuada, o bien suspender su uso cuando esto no sea posible. Es una actividad de responsabilidad compartida entre todos los agentes implicados en el uso del medicamento: el titular de la autorización de comercialización, las autoridades sanitarias, el médico, el farmacéutico, el enfermero y el paciente, así como el evaluador de las notificaciones de sospecha de reacciones adversas8. Con el objetivo de definir las bases que establecieran un sistema de calidad, la Agencia Europea del Medicamento (EMA) creó EudraVigilance en 2001, una red de procesamiento de datos para evaluar los acontecimientos adversos (AA) notificados por parte de las agencias regulatorias y compañías farmacéuticas en toda Europa.
La legislación en materia de farmacovigilancia está evolucionando de manera constante. Por otro lado, la planificación de la gestión de riesgos y las actividades relacionadas pueden variar entre los distintos países o regiones para poder adaptarlas al marco legal de cada país. Por ello, la UE creó un «sistema de gestión de riesgos» que engloba todas las actividades dedicadas a caracterizar y minimizar los posibles riesgos relacionados con un medicamento. En él se obliga a todo solicitante de autorización de un medicamento a presentar un Plan de Gestión de Riesgos (PGR), cuyo objetivo es garantizar la seguridad del paciente, del cual pueden requerirse actualizaciones en cualquier momento, si se considera necesario9.
En el caso de los productos biológicos, debido a sus peculiares características, todas estas actividades se deben realizar de manera más intensiva. De hecho, se incluyen como «grupo de medicamentos sujetos a seguimiento adicional» todos aquellos productos biológicos autorizados a partir del año 2011, grupo que pueden abandonar al igual que otros fármacos, transcurrido el tiempo necesario. Más concretamente, y debido al complejo proceso de producción, pequeñas variaciones en el mismo pueden afectar al producto final, incluso en productos que contienen la misma sustancia activa. Algunos ejemplos de ello son los AA con eritropoyetinas ocurridos en 200210,11, diferencias que se han notificado en el riesgo a desarrollar inhibidores del factor viii dependiendo de si el producto usado es de segunda o tercera generación12 o el incremento en el desarrollo de trombopatías microangiopáticas en pacientes tratados con formulaciones de interferón beta que presentaban pequeñas variaciones, como es el caso de Rebif® y Avonex®. Esto hace indispensable disponer de sistemas capaces de detectar cualquier diferencia potencial entre los diferentes productos biológicos, especialmente después de los cambios producidos en los procesos de fabricación, siendo necesario saber con precisión el lote de cada producto además de su nombre comercial (fig. 1).
. * Los ejemplos expuestos son solo a título ilustrativo. Recordar que Remsima e Inflectra son el mismo producto. Modificada de Vermeer et al.48.")
Variabilidad en el perfil de seguridad de los productos biológicos más allá del nivel de la sustancia activa.
ATC: clasificación anatómica terapéutica; INN: international nonproprietary name o denominación común internacional (DCI).
* Los ejemplos expuestos son solo a título ilustrativo. Recordar que Remsima e Inflectra son el mismo producto.
Modificada de Vermeer et al.48.
La llegada de los primeros productos biológicos a la práctica clínica en Europa trajo consigo especiales medidas de farmacovigilancia cuya eficacia ha sido contrastada. Sin embargo, su número ha crecido mucho en los últimos años, debido principalmente al gran número de nuevas moléculas innovadoras, pero también como consecuencia de la entrada en escena de los productos biosimilares (tabla suplementaria). Esto ha generado una situación de controversia en torno al perfil de seguridad y la eficacia de los nuevos biológicos, acompañada de continuos debates en torno a ellos e incluso en torno a sus productos originales13-16. Todo ello podría evitarse garantizando el cumplimiento de las leyes vigentes, así como facilitando una adecuada formación a todas las partes implicadas en su utilización.
Los métodos disponibles para evaluar la seguridad de los medicamentos biológicos, una vez comercializados, son muy diversos. El recurso más conocido entre los profesionales es el Sistema de Notificación Espontánea, basado en la notificación de AA surgidos en la práctica clínica rutinaria. En la actualidad, casi todos los países del mundo disponen de sistemas que permiten la notificación de AA tanto a los profesionales de la salud como a los propios pacientes17. Sin embargo, muy pocos AA son reportados y la calidad de las notificaciones puede variar, habiendo muchos casos en los que no podemos relacionar un determinado AA con el fármaco responsable sin temor a error. De hecho, la notificación de eventos relacionados con fármacos de manera voluntaria por parte del personal institucional no es del todo fiable y su grado de eficiencia varía mucho de un hospital a otro, como demuestran varios estudios18,19. Como dificultad añadida está el hecho de que, a pesar de que la ley vigente obliga a asegurar la trazabilidad de los medicamentos biológicos utilizados, el grado en el que esto se lleva a cabo varía según el centro sanitario en el que nos encontremos20.
Todo ello está muy en relación con el nivel de acceso a la información disponible. Así, para aquellos AA que tardan relativamente poco tiempo en aparecer, la información necesaria se puede obtener del envase del producto, pero si ya no se dispone de él, la calidad de la notificación va a depender de cada situación específica. En algunos casos, por ejemplo, el médico que se enfrenta al AA puede no ser el que ha prescrito el medicamento, siendo posible que no tenga acceso a toda la información necesaria para realizar un juicio clínico correcto. En otros casos, el paciente puede tener escasa capacidad para diferenciar los AA producidos por el fármaco de los síntomas de su propia enfermedad.
Otro recurso utilizado son las bases de datos electrónicas de los sistemas de salud (bases de datos médicas, de reclamaciones o de registros de medicación)21. Estas tienen la ventaja de asegurar el almacenamiento rutinario y sistemático de los datos clínicos. Su disponibilidad va a depender del grado de desarrollo y del adecuado funcionamiento de los registros farmacéuticos.
Por último, cabe destacar las bases de datos y los registros disponibles en las diferentes asociaciones médicas y sociedades científicas, que cada vez van ganando más importancia. Estas varían desde pequeños registros nacionales hasta grandes bases de datos a nivel internacional que trabajan con múltiples brazos de tratamientos y se focalizan en un amplio rango de escenarios clínicos. La mayoría de estos registros se basan en cohortes a largo plazo seleccionadas de las que se recoge información sobre los tratamientos, así como sobre logros clínicos obtenidos, todo ello a intervalos de tiempo predefinidos. Es el caso del Registro español de AA de terapias biológicas en enfermedades reumáticas o cutáneas (BIOBADASER, BIOBADADERM, respectivamente), el Británico de Reumatología (BSRBR)22,23 o del registro Europeo PedNet de Hemofilia24, por poner solo algunos ejemplos.
En la página web de la AEM podemos encontrar toda la información referida al Sistema Español de Farmacovigilancia, el cual integra las actividades que las administraciones sanitarias realizan en esta materia y está constituido por los Centros Autonómicos de Farmacovigilancia y la División de Farmacoepidemiología y Farmacovigilancia de la AEM, del Ministerio de Sanidad y Consumo así como por los profesionales sanitarios25.
Trazabilidad de los fármacos biológicosLa trazabilidad es un aspecto fundamental dentro del campo de la Farmacovigilancia. Por ella, entendemos la capacidad para reconstruir la historia, recorrido o aplicación de un producto farmacéutico, identificando para ello el origen de sus componentes, los procesos aplicados al mismo y la distribución y la localización desde su producción y durante todo su «ciclo vital». Consiste en poder identificar las etapas que un producto va superando dentro de su proceso de fabricación (manipulaciones, composición, maquinaria empleada, temperatura a la que está sometido, número de lote, etc.) que se consideren de importancia y que pueden hacer variar el producto final para el consumidor. Un sistema de trazabilidad adecuado deberá ser implementado por la totalidad de las personas físicas o jurídicas que intervengan en la cadena de producción, comercialización, distribución y dispensación del medicamento en cuestión (laboratorios, distribuidoras, operadores logísticos, farmacéuticos, médicos y enfermeras)26.
El fabricante deposita, en el envase de cada una de las unidades de venta al público, un soporte o dispositivo con capacidad para almacenar un código único e inequívoco. Esto permite identificar cada unidad del medicamento como única, lo cual es imprescindible para construir una cadena de seguimiento. También es imprescindible disponer de un sistema de captura de datos en el cual se puedan ir añadiendo todos los datos que se tengan disponibles sobre el medicamento hasta el momento actual y el cual sea accesible de manera sencilla y rápida27.
En 2013, las autoridades europeas promulgaron una ley (Directiva 62/2011) de medicamentos falsificados para todos los agentes de la cadena de distribución, según la cual, a partir del 2016, será obligatorio que cada producto contenga un código de barras individual que será almacenado en los registros del fabricante y posteriormente chequeado en los registros de dispensación. El sistema escogido para asegurar la trazabilidad de estos productos ha sido el Data Matrix, descartándose así otros procedimientos de lectura que también fueron analizados, como el de radiofrecuencia. El Data Matrix, que no compartirá espacio en el envase con ningún otro código identificativo, incluirá información sobre: ID del producto, número de serie, código nacional, número de lote y fecha de caducidad28.
Los fármacos biológicos son dispensados a través de varios sistemas de distribución en los que la información es recogida escaneando el código de barras del envoltorio exterior, que almacena la información que acompaña al nombre comercial del producto, el cual es único para cada fabricante y dosis. Esto permite un almacenamiento automático de toda la información del mismo29. Sin embargo, el lote del producto debería ser apuntado manualmente en cada dispensación cuando se realice mediante códigos de barras informáticos que, por el momento, no están incluidos. En las ocasiones en las que no hay un sistema informático disponible para la dispensación, toda esta información es almacenada manualmente (ya que es necesaria para muchas actividades administrativas que se requieren a la hora de manejar las existencias). En un estudio reciente, se llegó a la conclusión de que en la mayoría de los fármacos biológicos administrados en los hospitales sí se anotaba el lote, al contrario de lo que ocurre en las oficinas de farmacia de calle. Esto se debe principalmente a las leyes de buena preparación y a que la «Guía de productos medicinales derivados del plasma» obliga a la anotación del lote de todos los productos que deriven de plasma o de sangre30.
En la mayoría de casos, la administración de los medicamentos biológicos tiene lugar en los hospitales, debido a los complejos procesos que implican tanto su preparación como su administración, así como la necesaria monitorización que se les realiza posteriormente. La información recogida sobre la medicación va a depender, entre otros, de si los registros se realizan en papel o de forma electrónica, la correcta relación entre el circuito farmacia-administración y los protocolos de cada hospital sobre la recolección de la información según el tipo de biológico utilizado. Sin embargo, a este último respecto, hay algún producto que requiere condiciones especiales independientemente de los protocolos internos del hospital, como el caso de los hemoderivados o en el de infliximab y sus biosimilares, en cuyas guías europeas se recomienda la anotación del lote y la fecha de caducidad en el momento de la dispensación, tanto por parte de las administraciones sanitarias como del paciente31.
Afortunadamente, cada vez existen más iniciativas para mejorar los sistemas de trazabilidad. Francia ha sido el primer país en la UE en incluir una norma que obliga a la inclusión en la información de los Data Matrix32. Otros países, como Italia, Grecia y Bélgica, están tomando medidas en la misma dirección33. Las previsiones futuras indican que los avances en la tecnología permitirán la anotación automática tanto del lote como de la fecha de caducidad de cada producto individual34,35. Incluso está previsto el desarrollo de aplicaciones para móviles para cuando los pacientes se administren el medicamento en su domicilio36.
Retos e incertidumbres de los tratamientos biológicosIntercambiabilidadSeleccionar la terapia biológica adecuada y determinar si es necesario intercambiar un tratamiento biológico por otro equivalente implica tomar una decisión delicada en pacientes ya sea por ineficacia o por la aparición de AA debidos al tratamiento previo. La llegada de los fármacos biosimilares trae consigo la aparición de terapias más baratas e igual de eficaces, pero también plantea una duda en cuanto a la idoneidad o no del intercambio de un producto por otro.
Ante la ausencia de biomarcadores concretos para muchas enfermedades, es muy difícil establecer características universales que se asocien de manera inequívoca a conceptos como «fallo en el tratamiento» o «respuesta inadecuada». Así, en el caso de la AR, reumatólogos europeos y americanos han llegado a un consenso respecto a la definición de «remisión de la enfermedad» que repercute directamente en el manejo del paciente37. Además, los AA asociados a las terapias biológicas, como las reacciones infusionales o las complicaciones derivadas de los complejos sistemas de administración que muchas veces poseen estos productos, pueden producir la frustración del paciente haciendo que se considere que una terapia es ineficaz antes de tiempo. Un estudio publicado recientemente, realizado antes de la entrada en escena de los fármacos biosimilares, ha revelado que una tercera parte de los pacientes incluidos presentó más de un intercambio en el tipo de terapia biológica utilizado en un periodo de tan sólo 15 meses38.
Para asegurar, por tanto, una correcta intercambiabilidad, los datos de la terapia han de ser recogidos de manera continua. Sin embargo, basarse en estudios observacionales de seguridad para identificar diferencias significativas en eventos que ocurren con poca frecuencia, tales como reacciones infusionales o infecciones severas, no parece del todo fiable. Hay que tener en cuenta que estos productos también son utilizados en países con alta prevalencia de infecciones graves, como la tuberculosis, pudiendo sesgar la interpretación de los datos observacionales sobre seguridad en eventos infecciosos. Además, el periodo en el cual hacer el «switching» debe ser tenido muy en cuenta ya que puede ser determinante. Así, en pacientes que pierden la respuesta al tratamiento por la aparición de anticuerpos antifármaco, se debería realizar un cambio entre tratamientos39.
Otro aspecto a recordar es que los fármacos biológicos pueden presentar modificaciones que les lleven por derroteros independientes, tanto en el proceso de fabricación, de formulación, el acondicionamiento primario o incluso en la aparición de nuevas indicaciones terapéuticas. Esto puede crear un escenario en el cual las diferencias que se puedan producir entre los lotes del innovador o entre los diferentes biosimilares del mismo producto original se pueden incrementar en el tiempo40. Tener en cuenta esto a la hora de asegurar la biosimilitud es un gran reto para poder realizar un intercambio con garantías suficientes, ya que hasta el momento disponemos de poca evidencia científica en este terreno. Por todo ello, creemos que este nuevo escenario en la oferta terapéutica de biológicos a raíz de la aparición de los biosimilares implica el reto de afianzar todos los conocimientos que se tienen de estos medicamentos por parte de los agentes implicados en su manejo con el fin no comprometer la eficacia ni la seguridad que han presentado hasta la actualidad41.
Inmunogenicidad y su implicación en la calidad, eficacia y seguridadUno de los mayores problemas que se plantean con estos productos es que pueden existir diferencias en cuanto a la dinámica de formación de anticuerpos al compararlos con su producto de referencia o innovador. Así, en el caso de adalimumab, la aparición de anticuerpos ha sido relacionada con un incremento en la aparición de episodios tromboembólicos42. Sin embargo, no hay evidencia de que intercambios repetidos induzcan per se inmunogenicidad, por lo que cada caso hay que verlo de manera individual43. En el supuesto de infliximab, la mayoría de anticuerpos que actúan lo hacen uniéndose a su parte murina, mientras que los que aparecen contra adalimumab lo hacen uniéndose a la región determinante de complementariedad42.
La formación de anticuerpos frente a estos medicamentos puede tener diferentes mecanismos: reacciones de hipersensibilidad, reacciones cruzadas entre los receptores Fc (que pueden dar reacciones en el lugar de inyección) o reacciones de autoinmunidad inmediata no relacionadas con la formación de anticuerpos, lo cual ocurre con casi todos los anticuerpos monoclonales comercializados. Una excepción es el cetuximab, en el que anticuerpos antigalactosa-α-1,3-galactosa preexistentes han sido relacionados con reacciones infusionales fatales en pacientes que lo recibían por primera vez42. Por ello, este tipo reacciones no deben representar un obstáculo a la hora de realizar un intercambio, a lo que hay que añadir que los fabricantes de los biosimilares de este producto están utilizando menores cantidades de glicanos «no humanos». Por ello, es de esperar que estos produzcan menos reacciones infusionales, manteniendo la misma eficacia que el fármaco innovador. Esto crea a una situación paradójica, ya que en ocasiones el biosimilar podría ser menos inmunogénico que el producto innovador y aun así puede no realizarse el intercambio por temor a reacciones de inmunogenicidad.
Pese a todo, y apoyándonos en la escasa bibliografía disponible hasta la actualidad, se ha comprobado que el riesgo de inmunogenicidad es relativamente bajo después de 12 meses de tratamiento con infliximab. Solo el 90% de los pacientes que desarrolló anticuerpos de manera sostenida lo hizo dentro de los primeros 12 meses de tratamiento44. Lo mismo ocurre en el caso del adalimumab45.
Finalmente, hay que tener cierta cautela al manejar estos datos, utilizándolos de un modo comparativo y complementario. También se debe tener en cuenta la variabilidad de los resultados en función de la técnica diagnóstica utilizada44. Es decir, si un cambio potencial en la inmunogenicidad después de 12 meses de tratamiento fuera el mayor incidente acontecido, para averiguar su relevancia clínica se requeriría de un laboratorio centralizado así como la correlación de los resultados con los niveles del medicamento en el organismo, respuesta al tratamiento e incidencia y severidad de las reacciones adversas.
Nomenclatura y etiquetadoDado que sus peculiaridades moleculares dificultan la existencia de 2 copias exactas de estos fármacos, los productos biológicos deben poseer una nomenclatura que los defina e identifique inequívocamente. Como indica la EMA46, el etiquetado del medicamento, y en particular la «ficha técnica», es un aspecto fundamental a la hora de su comercialización en la UE y constituye la base de la información para los profesionales de la salud para utilizarlo de forma efectiva y segura. La ficha técnica está en continua actualización, reflejando cualquier cambio que se produzca durante todo el «ciclo vital» del producto. Sin embargo, en el caso concreto de los biológicos no está todavía claro qué información debe incluir. De hecho, en la última reunión de expertos en productos biosimilares se presentaron varias alternativas, sin que ninguna de ellas lograra una aceptación mayoritaria47.
La Organización Mundial de la Salud (OMS) establece un sistema de nomenclatura para los medicamentos, el «International Nonproprietary Name» o la «Denominación Común Internacional» (DCI), que es indicativo del componente activo y del grupo terapéutico. Este es reconocido de manera global y facilita la identificación inequívoca de las sustancias activas47.
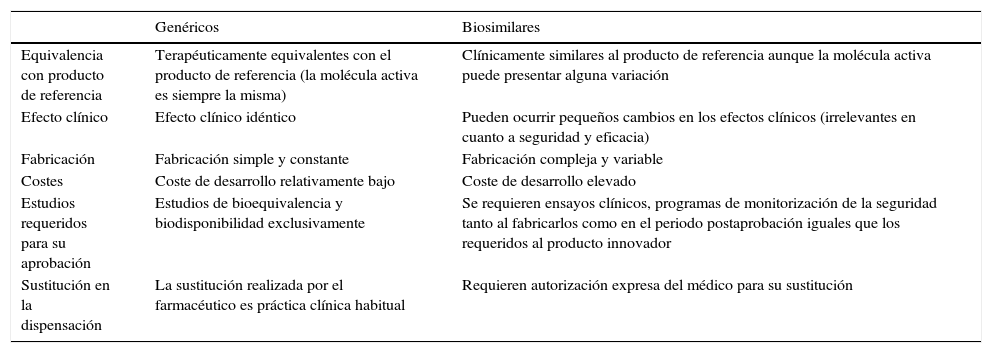
En el caso de los biosimilares, como de los genéricos, los medicamentos deberían utilizarse solo en las indicaciones autorizadas, y excepcionalmente fuera de ficha técnica cuando no hay otras alternativas. Sin embargo, el biosimilar, a diferencia del genérico, no puede utilizarse automáticamente en todas las indicaciones del producto de referencia, requiriendo cualquier extrapolación de ellas una justificación especial (tabla 2).
Diferencias entre los fármacos genéricos y los fármacos biosimilares con sus correspondientes productos de referencia
| Genéricos | Biosimilares | |
|---|---|---|
| Equivalencia con producto de referencia | Terapéuticamente equivalentes con el producto de referencia (la molécula activa es siempre la misma) | Clínicamente similares al producto de referencia aunque la molécula activa puede presentar alguna variación |
| Efecto clínico | Efecto clínico idéntico | Pueden ocurrir pequeños cambios en los efectos clínicos (irrelevantes en cuanto a seguridad y eficacia) |
| Fabricación | Fabricación simple y constante | Fabricación compleja y variable |
| Costes | Coste de desarrollo relativamente bajo | Coste de desarrollo elevado |
| Estudios requeridos para su aprobación | Estudios de bioequivalencia y biodisponibilidad exclusivamente | Se requieren ensayos clínicos, programas de monitorización de la seguridad tanto al fabricarlos como en el periodo postaprobación iguales que los requeridos al producto innovador |
| Sustitución en la dispensación | La sustitución realizada por el farmacéutico es práctica clínica habitual | Requieren autorización expresa del médico para su sustitución |
Con el fin de evitar el error que esto puede producir a la hora de prescribir un biosimilar en una indicación en la que no esté aprobado, la OMS ha propuesto añadir una serie de números y letras que siguen al DCI mediante prefijos y sufijos clasificando las moléculas de una forma más precisa. Además, para los biológicos que tengan proteínas con residuos glicosilados de manera extensiva, establece el uso de sufijos adicionales. De este modo, los biológicos adquieren nombres singulares y «únicos». Este sistema ha tenido por ahora diferente grado de aceptación a nivel mundial. De hecho, la EMA no se ha acogido por el momento a él47.
Sin embargo, y a pesar de estas medidas, se sigue utilizando el mismo nombre para diferentes productos (con distintos fabricantes, procesamiento, formulación, sistemas de administración, etc.)48. Ejemplo de ello es el «interferón beta-1». Recientemente, los fabricantes de los biológicos innovadores propusieron establecer un sistema adicional al DCI que advirtiera que determinado medicamento es un biosimilar, ante lo cual los fabricantes de biosimilares recuerdan que en la actualidad los biológicos innovadores conservan siempre el mismo DCI a pesar de presentar variaciones en sus restos glicosilados, siendo esto contradictorio ya que productos aprobados como altamente similares poseen diferente nombre49.
Normas regulatoriasLlegados a este punto, es conveniente recordar que tanto el producto biológico innovador como el biosimilar siguen un procedimiento obligado de registro centralizado que es coordinado por la EMA. El medicamento de referencia con el que se compara el biosimilar debe estar autorizado en la UE y debe ser el mismo para todo el programa de desarrollo del biosimilar. En la EMA existen grupos de expertos en biológicos, como el Biologics Working Party, y en biosimilares en particular como el Biosimilar Medicinal Products Working Party, que se encargan de dar opinión en todo lo referente a estos medicamentos para que el Committee for Medicinal Products for Human Use se encargue de redactar las guías de medicamentos biológicos, emitiendo una opinión científica sobre la aprobación y en base a ello la Comisión Europea (CE) tome la decisión final sobre la autorización de comercialización en la UE50. Este procedimiento centralizado es válido en todos los países miembros de la UE. En los últimos años, la EMA ha publicado varias guías en las que describe las condiciones de autorización tanto de los biológicos innovadores como de los biosimilares51-53.
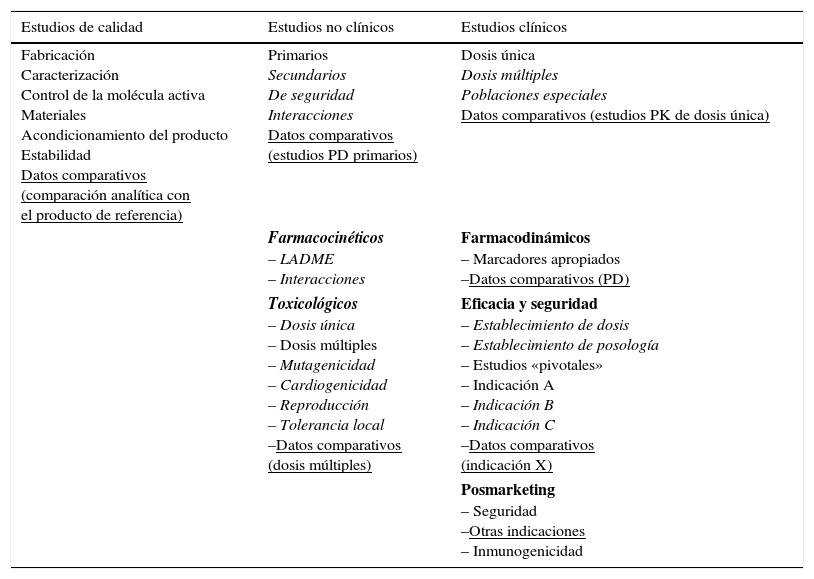
Por otro lado, al desarrollar un biosimilar se pretende demostrar la semejanza entre el biosimilar y el fármaco original en cuanto a la eficacia y la seguridad previamente justificadas. Por ello, los procedimientos requeridos para su aprobación son diferentes pero igual de estrictos (tabla 3). Esto permite reducir el coste económico y acelerar su aparición en el mercado. Al igual que para el resto de los medicamentos, cada empresa farmacéutica debe presentar un PGR junto con la solicitud de autorización de comercialización. Todos los medicamentos biosimilares del mercado disponen de un PGR con información sobre el plan incluidos en el informe de evaluación publicado en el sitio web de la EMA. El PGR de un medicamento biosimilar debe considerar el perfil de seguridad conocido del medicamento de referencia. Al ser la inmunogenicidad uno de los principales temas de interés de cualquier medicamento biológico, esto debe abordarse en el informe PGR, pudiendo requerirse estudios de eficacia postautorización (en inglés: PAES), si así lo decide la CE, por ejemplo, cuando existan preocupaciones relativas a determinados aspectos de eficacia o seguridad que solo puedan resolverse una vez que este se haya comercializado54.
Requerimientos de la EMA para la aprobación de los productos biológicos innovadores y de los biosimilares
| Estudios de calidad | Estudios no clínicos | Estudios clínicos |
|---|---|---|
| Fabricación Caracterización Control de la molécula activa Materiales Acondicionamiento del producto Estabilidad Datos comparativos (comparación analítica con el producto de referencia) | Primarios Secundarios De seguridad Interacciones Datos comparativos (estudios PD primarios) | Dosis única Dosis múltiples Poblaciones especiales Datos comparativos (estudios PK de dosis única) |
| Farmacocinéticos – LADME – Interacciones | Farmacodinámicos – Marcadores apropiados –Datos comparativos (PD) | |
| Toxicológicos – Dosis única – Dosis múltiples – Mutagenicidad – Cardiogenicidad – Reproducción – Tolerancia local –Datos comparativos (dosis múltiples) | Eficacia y seguridad – Establecimiento de dosis – Establecimiento de posología – Estudios «pivotales» – Indicación A – Indicación B – Indicación C –Datos comparativos (indicación X) | |
| Posmarketing – Seguridad –Otras indicaciones – Inmunogenicidad | ||
Letra normal: requerimientos habituales para ambos productos; en cursivas: requerimientos exclusivos para el producto de referencia; subrayado: requerimientos exclusivos para los biosimilares.
LADME: liberación, absorción, distribución, metabolismo y excreción; PD: farmacodinámicos; PK: farmacocinéticos.
Por lo general, los fármacos biológicos son más costosos que los medicamentos de pequeña molécula y gestionar correctamente su uso es un objetivo cada vez de mayor importancia para los organismos pagadores. Los biosimilares pueden ofrecer una alternativa más económica que los productos biológicos existentes que han perdido ya sus patentes y favorecer una limpia competencia, mejorando por tanto el acceso de un mayor número de pacientes a los medicamentos biológicos, contribuyendo así a la sostenibilidad financiera de los sistemas sanitarios. Aunque la diferencia de precios entre los biosimilares y sus correspondientes medicamentos de referencia en valores absolutos no tiene todavía el impacto de los fármacos genéricos, los biosimilares han experimentado un crecimiento en los últimos años, reduciendo de forma clara el coste de las terapias biológicas55.
ConclusionesLos sistemas de farmacovigilancia se basan en la monitorización y la evaluación de toda la información relacionada con un fármaco con el fin de evaluar su seguridad una vez comercializado. La trazabilidad es una herramienta indispensable dentro de los mismos, ya que nos permite reconstruir la historia de un medicamento durante todo su «ciclo vital», pudiendo detectar los acontecimientos, tanto intrínsecos como extrínsecos al fármaco que puedan hacer variar las características del producto que llega al destinatario final, el paciente.
Para la notificación de reacciones adversas a medicamentos (RAM) relativas a cualquier fármaco biológico, innovadores y biosimilares, es de vital importancia identificar claramente el medicamento del que se trata. Así, la legislación europea obliga a que toda notificación de una reacción adversa a un medicamento biológico vaya acompañada del nombre aprobado del medicamento y del número de lote, hecho que debe ser garantizado por las autoridades locales de cada estado miembro de la UE56. Como vemos, la trazabilidad cobra especial interés en el campo de los fármacos biológicos, y así lo reconocen las autoridades competentes.
En este trabajo, hemos revisado los aspectos principales relacionados con ambos procesos, aplicados al mundo de los productos biológicos. Asimismo, hemos abordado conceptos como intercambiabilidad, inmunogenicidad, nomenclatura y las normas que regulan el uso de estos productos. Finalmente, creemos que este es un campo apasionante para el clínico y en continua expansión, sujeto a nuevos conceptos y cambios constantes provenientes tanto de la ciencia básica, como de la industria farmacéutica, la práctica clínica y las administraciones sanitarias, debiendo existir una correcta interacción entre todas ellas para poder alcanzar los objetivos más adecuados y coste-eficientes.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener conflictos de interés en relación con el presente trabajo.
Los autores agradecen a todos los integrantes del Servicio de Farmacia del Hospital de La Princesa por los comentarios y las críticas constructivas que han ayudado a mejorar este trabajo.


