




El síndrome de Sjögren primario (SSp) es una enfermedad sistémica autoinmune, de progresión lenta y etiología desconocida, que se caracteriza por la infiltración linfocitaria de las glándulas exocrinas, provocando especialmente xeroftalmia y xerostomía. Puede afectar a cualquier órgano o sistema, provocando manifestaciones extraglandulares, que incluso pueden preceder a las típicas manifestaciones glandulares y retrasar el diagnóstico. En los últimos años el mayor conocimiento de la enfermedad ha inducido también mejoras en su manejo terapéutico.
Primary Sjögren′s syndrome (pSS) is a chronic systemic autoimmune disease, of slow progression, characterized by lymphocytic infiltration of the exocrine glands, that leads to sicca symptoms, mainly xerophtalmia and xerostomia. It may involve any organ and lead to extraglandular manifestations, which also can precede typical glandular manifestations and delay the diagnosis of pSS. In the past years, better knowledge of the disease has led to improvement in treatment management.
El síndrome de Sjögren (SS) es una exocrinopatía crónica autoinmune, de progresión lenta y etiología desconocida. También se le denomina epitelitis autoinmune, por ser el epitelio de las glándulas exocrinas las células diana de la respuesta inflamatoria provocada por la infiltración linfoplasmocitaria, presencia de autoanticuerpos y mediadores de la inflamación. Se considera al SS como primario (SSp) si aparece de forma aislada y secundario si se presenta asociado a otra enfermedad autoinmune como el LES o la artritis reumatoide1,2. Se caracteriza principalmente por la sequedad de la mucosa bucal (xerostomía) y ocular (xeroftalmia), si bien con frecuencia puede producir síntomas por sequedad nasal, cutánea o vaginal. Aunque en la mayoría de los pacientes la enfermedad suele quedar localizada en las glándulas exocrinas (manifestaciones glandulares), la reacción inflamatoria puede afectar de forma sistémica a diversos órganos y provocar manifestaciones extraglandulares. En este artículo analizaremos las principales manifestaciones extraglandulares en los pacientes con SSp y su manejo.
Manifestaciones extraglandularesEn general, el 70% de los pacientes con sospecha de SSp, son derivados para evaluación a las consultas de reumatología por la presencia de síntomas de sequedad de mucosas (xerostomía, xeroftalmia, etc.), en un 20% por presentar anticuerpos y solo alrededor del 10% por alguna afección extraglandular.
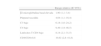
Las manifestaciones extraglandulares son diversas y varían según las series utilizadas. En ocasiones pueden ser la forma de presentación de la enfermedad o detectarse antes que las manifestaciones glandulares típicas y retrasar el diagnóstico del propio SSp. La serie española publicada por Ramos-Casal et al3, que incluye a 1.010 pacientes con SSp, es hasta la fecha la más amplia, será la que se utilizará en este artículo, comentando las que se presenten en más del 5% de los casos. Otras series internacionales incluyen menos pacientes y muestran discretas diferencias dependiendo de los criterios utilizados4,5. A lo largo de la evolución de la enfermedad, pueden aparecer hasta en el 50% de los pacientes con SSp, por la presencia de artralgias. Sin embargo, al excluir este síntoma, la prevalencia de manifestaciones extraglandulares alcanzaría al 20% de los pacientes (tabla 1).
Manifestaciones extraglandulares en el síndrome de Sjögren primario (adaptado de Ramos-Casals M, et al Medicine 20083)
| Manifestaciones extraglandulares | |
| Artralgias | 48% |
| Fenómeno de Raynaud | 18% |
| Artritis | 15% |
| Sistema nervioso | 13% |
| SN periférico | 11% |
| SN central | 2% |
| Pulmonar | 11% |
| Vasculitis | 9% |
| Renal | 5% |
| Alteraciones inmunológicas principales | |
| ANA | 85% |
| Anti-Ro | 52% |
| Factor reumatoide | 48% |
| Anti-La | 34% |
| Crioglobulinas | 10% |
| C3 bajo | 9% |
| C4 bajo | 9% |
Las artralgias son la manifestación extraglandular más frecuente, afectando al 48% de los pacientes3. La artritis, habitualmente oligoarticular e intermitente, afecta con mayor frecuencia a manos y rodillas, se detecta en el 15%. Aunque puede provocar deformidad articular tipo Jaccoud, a diferencia de la artritis reumatoide no provoca erosiones. El factor reumatoide está presente entre el 50 al 75% de los casos3,6. Sin embargo, los anti-péptidos cíclicos citrulinados se detectan en menos del 10% de los pacientes7. En raras ocasiones se detecta la presencia de miopatía, con debilidad muscular proximal y discreta elevación de las enzimas musculares.
El tratamiento sintomático con AINE puede ser útil. En los pacientes con artritis se aconseja probar la hidroxicloroquina y si no hay respuesta valorar la introducción de metotrexato en monoterapia o asociado a hidroxicloroquina. Algunos autores plantean el uso prolongado de hidroxicloroquina en los pacientes con SSp, dado que interfiere en el reconocimiento de antígenos e inhiben el interferón gamma8 y en un estudio controlado con placebo disminuyó de forma significativa la VSG y la hipergammaglobulinemia IgG e IgM9, aunque se desconoce en la actualidad si esta pauta puede disminuir la aparición de otras manifestaciones de la enfermedad.
Fenómeno de RaynaudEl fenómeno de Raynaud se detecta en el 18% de los pacientes con SSp3. En cerca de la mitad de los casos incluso precede al diagnóstico. Se asocia de forma significativa a la presencia de síntomas articulares, vasculitis cutánea, ANA, anti-Ro y anti-La10. Aunque es infrecuente la aparición de complicaciones isquémicas graves, su presencia suele estar relacionada con vasculitis con crioglobulinas. El tratamiento suele ser sintomático con fármacos vasodilatadores dependiendo de la intensidad de la clínica.
TiroidesLa afectación tiroidea es frecuente en los pacientes con SSp. Se atribuye a que tanto las glándulas salivales como la glándula tiroidea comparten características histológicas y antigénicas. Aunque en algunas series se detectan alteraciones tiroideas hasta en el 70% de los pacientes con SSp, en un estudio de casos y controles bien diseñado, la afectación tiroidea fue del 36% y no se observaron diferencias tanto en las alteraciones tiroideas inmunológicas (20%) como en las no inmunológicas (16%)11. Entre los pacientes con SSp, la enfermedad tiroidea solo se relacionó de forma significativa con la presencia de anticuerpos antiroideos antiroglobulina y peroxidasa. Suele ser clínicamente silente y aparecer como hipotiroidismo subclínico.
Vasculitis cutáneaLa vasculitis cutánea aparece en el 10% de los pacientes con SSp3. La vasculitis leucocitoclástica de pequeño vaso es el patrón histológico más frecuente (95%). Suele presentarse en la mayoría como púrpura palpable no asociado a crioglobulinas y con menor frecuencia asociado a vasculitis crioglobulinémica y a vasculitis urticarial. Suelen ser pacientes con gran expresión clínica sistémica y presencia de otras manifestaciones extraglandulares de la enfermedad. Se relaciona de forma significativa con artritis, neuropatía periférica, fenómeno de Raynaud, glomerulonefritis, ANA, factor reumatoide, anti-Ro, aumento de hospitalización e incremento de la mortalidad3,12. La presencia de vasculitis, crioglobulinas y C4 bajo se consideran factores de mal pronóstico entre los pacientes con SSp, asociándose a la presencia de manifestaciones extraglandulares y aumento de la mortalidad6,12.
En pacientes con afectación leve o exclusivamente cutánea se puede mantener actitud expectante. Ante el deterioro clínico, presencia de crioglobulinas o de otras manifestaciones extraglandulares de la enfermedad, se deben de utilizar corticoides, con frecuencia asociados a inmunosupresores. En casos refractarios el rituximab es una opción a considerar13.
PulmonarLa afectación pulmonar se detecta en el 11% de los pacientes3, diferenciando: la vía aérea, el parénquima pulmonar y la vascularización (tabla 2).
Afectación pulmonar en el SSp
| 1. | Vía aérea |
| Sequedad vías aéreas | |
| Bronquitis/bronquilitis | |
| 2. | Enfermedad pulmonar intersticial (EPI) |
| Neumonía intersticial linfocítica | |
| Neumonía intersticial no-específica | |
| Neumonía intersticial usual | |
| 3. | Enfermedad linfoproliferativa |
| Infiltración linfocítica folicular: bronquial/bronquiolar | |
| Pseudolinfoma/linfoma | |
| Amiloidosis | |
SSp: síndrome de Sjögren primario.
Se refiere a la afectación del epitelio glandular de la mucosa de la vía aérea, por lo que se manifiesta por sequedad nasal, faríngea, laríngea, traqueal y bronquial. Es frecuente su afectación en estos pacientes. Se manifiesta clínicamente con tos, habitualmente poco productiva, por lo que puede confundirse con cuadros de rinitis alérgica o de bronquitis. La exploración, incluso con laringoscopia, suele ser poco expresiva o normal en la mayoría de los casos. El manejo terapéutico se basa en los tres pilares de la sequedad glandular: 1) Prevenir: uso de humificadores, evitar ambientes secos; 2) Sustituir: con hidratación abundante y spray nasal (agua de mar esterilizada) y bucal (saliva artificial), y 3) Estimular: en estos casos se puede probar con N-acetil cisteína oral.
Parénquima pulmonarLa enfermedad pulmonar intersticial (EPI), con diversas formas histológicas es la afectación más frecuente. Con mucha menor frecuencia puede desarrollar amiloidosis y linfoma.
Enfermedad pulmonar intersticialLa EPI puede ser la forma de presentación de la enfermedad, sin olvidar que el 20% de la EPI idiopática, está relacionada con la presencia de una colagenopatía, siendo el SSp el 8–10%14. Se considera que el pronóstico de la EPI es mejor si está en relación a una colagenopatía15. Estudios observacionales prospectivos indican que la aparición de la EPI en los pacientes con SSp suele ser precoz, en los primeros 4 años de la enfermedad16. En comparación con los pacientes con SS secundario, en los pacientes con SSp la EPI es más frecuente, aunque suele ser de menor gravedad. La presencia de derrame pleural es raro en los pacientes con SSp y debe hacer sospechar la existencia de linfoma.
La presencia de algunos factores han demostrado una alta especificidad para el desarrollo de EPI, señalando un perfil de paciente en el que habría que investigar la presencia de esta afección17: hipergammaglobulinemia (94%), linfopenia (93%), anti-Ro (86%), anti-La (93%), factor reumatoide (80%), descenso de la capacidad vital forzada (87%) o del volumen expiratorio en el primer segundo (92%, llegando al 100% en no fumadores) o el fenómeno de Raynaud (75%). Por otra parte, se ha demostrado de forma significativa que la edad, el sexo varón y el tabaquismo, son factores de riesgo para el desarrollo de EPI en pacientes con SSp17.
En la evaluación de pacientes con sospecha de EPI, se debe conocer el valor de las pruebas complementarias de uso habitual.
- 1.
Radiología de tórax
Suele ser el primer paso, pero presenta mala correlación clínica-histológica y puede ser normal o no detectarse alteraciones cuando ya el paciente las presenta en el TAC de alta resolución (TACAR) pulmonar. De hecho, en el estudio de Uffmann18, el 65% de los pacientes asintomáticos y con radiografía normal, presentaban alteraciones en el TACAR pulmonar. En otro estudio19, realizado en 59 pacientes con SSp, en el que el intervalo entre la radiografía y el TACAR era menor de 3 meses, el 50% de los pacientes presentaban alteraciones en el TACAR frente al 20% de las radiografías de tórax.
- 2.
Pruebas funcionales respiratorias
Suelen estar alteradas en estos pacientes. El patrón obstructivo indica afectación de la vía aérea y el patrón restrictivo afectación del parénquima pulmonar. Sin embargo, en muchas ocasiones podemos encontrar alteraciones mixtas. En el 20% de los pacientes asintomáticos con pruebas funcionales respiratorias normales se detectan alteraciones de EPI en el TACAR pulmonar18.
- 3.
Lavado bronquioalvolar
En los pacientes con SSp, predomina la presencia de linfocitos CD4 en el lavado bronquioalveolar, incluso de forma subclínica en el 50% de los casos. Se suele realizar en la evaluación de los pacientes con patrón intersticial y es de ayuda para el diagnóstico diferencial para descartar la presencia de neoplasias o infección. Algunos autores aconsejan realizarlo previamente al tratamiento inmunosupresor intenso como la ciclofosfamida.
La presencia de linfocitosis mayor del 15% (alveolitis de alto grado) se le relaciona con la presencia de sintomatología (tos, disnea), alteración de la difusión pulmonar (DLCO) y peor pronóstico a largo plazo. En un estudio realizado en pacientes con una media de seguimiento de 12 años20,21, solo los pacientes con linfocitosis basal mayor del 15%, precisaron durante la evolución tratamiento con corticoides (5 de 12 pacientes versus 0 de 10 pacientes; p<0,05) o hubo mortalidad (6 de 12 pacientes versus 0 de 10 pacientes; p<0,01). Sin embargo, no hubo diferencias en la evolución a EPI.
Las formas histológicas principales de la EPI en los pacientes con SSp son:
- 1.
Neumopatía intersticial no específica
Es la forma más frecuente de EPI, con una prevalencia del 60%. Presenta un buen pronóstico con una supervivencia a los 5 años del 83%. Existe buena correlación entre el resultado del TACAR pulmonar y el resultado histológico, con un valor predictivo positivo del 94%, que evita en muchas ocasiones la realización de una biopsia pulmonar22,23. Se presenta con imágenes reticulares basales en vidrio deslustrado24.
- 2.
Neumopatía intersticial usual
La prevalencia alcanza al 6% de los casos. Cursa de forma insidiosa con tos y disnea progresiva y no suele acompañarse de síntomas sistémicos. En el TACAR predomina la fibrosis con áreas en panal, con especificidad que alcanza el 80–90%24. En el lavado broncoalveolar, aunque la celuraridad puede ser normal, predominan los neutróficos con o sin eosinofilia.
- 3.
Neumopatía intersticial linfocítica
Es una proliferación bronquial linfoide benigna. Su prevalencia es del 1%. Aunque responde a corticoides, su pronóstico es peor, con una mortalidad a los 5 años del 50% de los pacientes. Se estima que el 5% evoluciona a linfoma15,22,23.
El tratamiento inicial de la EPI en los pacientes con SSp se basa en el uso de prednisona, a dosis de 1–2mg/kg/d, entre 1 a 2 meses para posteriormente reevaluar con controles de TACAR y pruebas respiratorias, especialmente de difusión o DLCO. Si ha habido respuesta se puede iniciar una pauta descendente con controles clínicos. Si no ha habido respuesta clínica (progresión en el TACAR o disminución mayor del 10% en DLCO), se aconseja introducir tratamiento inmunosupresor, inicialmente ciclofosfamida en bolus como inductor de la remisión25, con una pauta mensual similar al realizado en otras colagenopatías, para posteriormente continuar con azatioprina de mantenimiento26. Se han descrito casos con buena respuesta a rituximab, especialmente en pacientes con otras manifestaciones extraglandulares del SSp13,27.
Pseudolinfoma y linfomaEl denominado pseudolinfoma es un verdadero linfoma no Hodgkin (LNH) de bajo grado, de células B, tipo mucosa-associated lymphoid tissue (MALT)28. Aunque es el 1% de todos los LNH, a nivel pulmonar es el tumor más frecuente (65%). Con frecuencia provoca escasa sintomatología y suele ser un hallazgo radiológico casual. En el TACAR se puede apreciar la presencia de nódulos o consolidación uni/bilateral. El diagnóstico se realiza mediante biopsia pulmonar. El tratamiento se basa en la cirugía combinado a quimioterapia asociada o no a radioterapia29,30. El uso de rituximab parece atractivo y podría plantearse como tratamiento conminado a otras terapias en algunos pacientes.
Hipertensión pulmonarEs una complicación rara en los pacientes con SSp. Suele presentarse asociado a la presencia de fenómeno de Raynaud y EPI. Se aconseja iniciar el tratamiento con inmunosupresores y si no hay respuesta asociar vasodilatadores como el bosentán31.
Sistema nerviosoAunque la prevalencia varía según las series, la afectación del sistema nervioso (SN) periférico alcanza al 11% de los pacientes y a nivel del SN al 2%3. En este caso, la aparición de síntomas neurológicos suele preceder en el 40–80% de los casos al diagnóstico del SSp32,33.
El espectro de la afectación del SN en el SSp es amplio (tabla 3):
- 1.
Manifestaciones neuropsiquiátricas
Son frecuentes. La prevalencia de depresión es del 30% y la fatiga profunda alcanza al 65% de los pacientes. Sin embargo, casos de demencia son raros33.
- 2.
SN periférico
Se suelen acompañar de otras manifestaciones extraglandulares del SSp (especialmente vasculitis cutánea) y de presencia de alteraciones analíticas como hipergammaglobulinemia, linfopenia y crioglobulinas32,33.
La clínica principal es el dolor de tipo neuropático. Se pueden presentar como neuropatía sensitivomotora, neuropatía sensitiva pura no axonal (exclusiva del SSp) e incluso como vasculitis en forma de mononeuritis múltiple y neuropatías de pares craneales33,34.
- 3.
SN central
Con menor frecuencia aparecen cuadros en el SN central, como mielitis transversa, neuromielitis óptica, déficits focales similares a los que aparecen en el LES y en raras ocasiones vasculitis cerebral. Además se pueden detectar en la resonancia magnética cerebral, imágenes hiperintensas en la sustancia blanca cerebral habitualmente múltiples, redondeadas y de pequeño tamaño, que aparecen en la esclerosis multiple32,33,35,36. En los pacientes con SSp con frecuencia se pueden detectar estas imágenes inespecíficas en sujetos asintomáticos de significado incierto, puesto que pueden detectarse en pacientes con hipertensión arterial, migraña, depresión e incluso con la edad35,36.
Aunque el SSp puede coincidir con la esclerosis múltiple en menos del 3%, en ocasiones, puede ser difícil diferenciarlas. Ambas enfermedades predominan en mujeres de mediana edad, presentan bandas oligoclonales en el líquido cefalorraquídeo e incluso presentan imágenes en sustancia blanca cerebral por resonancia magnética. Sin embargo, la presencia de sintomatología glandular (xerostomía, xeroftalmia, etc.), manifestaciones sistémicas (vasculitis cutánea, pulmonar, etc.) o la detección de anticuerpos como anti-Ro o anti-La, señalan al SSp como diagnóstico32,33.
Afectación neurológica en el SSp
| 1. | Manifestaciones psiquiátricas |
| Depresión (30%) | |
| Fatiga: (65%) | |
| 2. | SN central (2%) |
| Déficits focales | |
| Mielitis transversa | |
| Neuromielitis óptica | |
| Vasculitis cerebral | |
| 3. | SN periférico (11%) |
| Neuropatía sensitivomotora | |
| Neuropatía sensitiva pura no axonal (exclusiva SSp) | |
| Mononeuritis múltiple | |
| Neuropatías pares craneales | |
SSp: síndrome de Sjögren primario.
No existe un tratamiento estandarizado en la afectación del SN en los pacientes con SSp. Sin embargo, algunos aspectos pueden ser de interés32,34,35:
- 1.
Control estricto de los factores de riesgo cardiovascular, especialmente la hipertensión arterial.
- 2.
En los pacientes asintomáticos, con lesiones en la sustancia blanca detectados por resonancia magnética se aconseja control evolutivo.
- 3.
En caso de progresión clínica o datos de actividad (déficit focal, mielitis transversa, encefalopatía), corticoides asociados a inmunosupresores, especialmente ciclofosfamida.
- 4.
En casos refractarios, se plantea el uso de inmunoglobulinas, plasmaféresis y rituximab.
Según la afectación del SN, algunos fármacos han demostrado mayor eficacia32,34,35:
- •
Corticoides: escasa eficacia en neuropatía axonal y no axonal.
- •
Ciclofosfamida en bolus iv: de elección en mononeuritis múltiple, mielitis transversa.
- •
Inmunoglobulinas: se plantea en la neuropatía sensitiva y es especialmente eficaz en casos de radiculoneuropatía (100%).
- •
Plasmaféresis: algunos casos de mielitis aguda y extensa.
- •
Rituximab: debe de plantearse en casos de neuromielitis óptica y de afectación del SN central desmielinizante.
La afectación renal aparece en el 5% de los pacientes, habitualmente de forma silente3. La infiltración linfocitaria puede afectar al glomérulo provocando glomerulonefritis membranosa o mebranoproliferativa. Sin embargo, con mayor frecuencia puede provocar nefritis intersticial, afectando a los túbulos, provocando acidosis tubular, con imposibilidad para acidificar la orina, no disminuyendo el ph urinario por debajo de 6 a pesar de la presencia de acidosis metabólica37. En casos avanzados existe hipokaliemia y tendencia a la formación de cálculos renales, precisando tratamiento con bicarbonato, e incluso corticoides.
LinfomaEl desarrollo de linfoma en los pacientes con SSp es la complicación más temida. El riesgo a lo largo de la vida es del 5%. Además, el riesgo aumenta con el tiempo de evolución de la enfermedad38: si en los primeros 5 años es de 6.4 (IC: 1,3–18,7), a los 10–15 años el riesgo de 20 (6-8-48,6). Suelen ser linfomas no Hodgkin de células B, de bajo grado y tipo MALT. El trabajo de Kassan et al publicado en 197839, demostró un riesgo de linfoma en estos pacientes de 44 veces. Sin embargo, estudios recientes, como el del grupo de Theander38, realizado en 500 pacientes con SSp y un seguimiento medio de 8 años, solo los pacientes diagnosticados cumpliendo los criterios de consenso Europeos-Americanos de 200240 (en los que es imprescindible para el diagnóstico, la presencia de biopsia labial positiva o de anti-Ro/anti-La), se demostró un riesgo aumentado de linfoma de cerca de 16 veces (tasa estandarizada de cáncer: 15,57; IC: 7,77–27,85), frente a la ausencia de casos de linfoma entre los pacientes diagnosticados según criterios europeos de 199341.
Algunos factores se han relacionado de forma significativa con el riesgo de linfoma6,38 (tabla 4): niveles elevados de beta2-microglobulina, presencia de vasculitis cutánea, niveles descendidos tanto de C3, C4 como de linfocitos CD4 o del ratio CD4/CD8 menor de 0.8.
Terapias en expectativaEl SSp se caracteriza por la hiperreactividad o expansión clonal de linfocitos B, tanto a nivel glandular como extraglandular. Gran parte de las terapias en investigación van dirigidas a intentar alterar o inhibir este aspecto esencial de la patogénesis. Para ello se están estudiando fármacos que actúan a distintos niveles42,43: 1) Inhibidores del interferón: desarrollando anticuerpos monoclonales. 2) Inhibidores del B-cell Activating Factor (BAFF), que se encuentra elevado en los pacientes con SSp; belimumab (anticuerpo monoclonal) o el BAFF-R-Ig (receptor soluble que se liga a BAFF) o el atacicept (receptor soluble), que se liga al BAFF y al A Proliferation-Inducing Ligand (APRIL). 3) Depleción de células B: rituximab (anti-CD20) y eprazutumab (anti-CD22). 4) Otros tratamientos: interleucinas IL6, IL21, que participan en activación de los linfocitos B; abatacept, que inhibe la presentación del antígeno a la célula epitelial; baminercept (receptor soluble beta); o el uso de linfotoxinas.


