




La artritis reumatoide (AR) es una enfermedad inflamatoria, sistémica y crónica de etiología desconocida y con predisposición genética. La llegada de los nuevos agentes biológicos, así como los ya conocidos fármacos antirreumáticos modificadores de la enfermedad, condujeron a una eficacia elevada en los tratamientos de la AR. Sin embargo, no todos los sujetos muestran el mismo grado de progresión de la enfermedad como respuesta a estos tratamientos. Estas variaciones demuestran que los sujetos con AR deben tener diferentes mecanismos de regulación génica. Los polimorfismos detectados en las regiones reguladoras no codificantes del sistema inmune y las variaciones genéticas de las enzimas que metabolizan los fármacos demuestran que este tipo de variaciones tiene una importancia funcional y evolutiva elevada, lo que proporciona nuevas pistas para el desarrollo de nuevas estrategias terapéuticas. La farmacogenética es un campo que avanza rápidamente y promete el desarrollo de tratamientos adaptados al perfil genético del sujeto en un futuro cercano.
Rheumatoid arthritis (RA) is a systemic, chronic and inflammatory disease of unknown aetiology with a genetic predisposition. The advent of new biological agents, as well as the more traditional disease-modifying anti rheumatic drugs, has resulted in highly efficient therapies for reducing the symptoms and signs of RA; however, not all patients show the same level of response regarding disease progression to these therapies. These variations suggest that RA patients may have different genetic regulatory mechanisms. The extensive polymorphisms revealed in non-coding gene-regulatory regions in the immune system, as well as genetic variations in drug-metabolizing enzymes, suggest that this type of variation is of functional and evolutionary importance and may provide clues for developing new therapeutic strategies. Pharmacogenetics is a rapidly advancing area of research that holds the promise that therapies will soon be tailored to an individual patient's genetic profile.
La artritis reumatoide (AR) es una enfermedad crónica, sistémica e inflamatoria que lleva a la destrucción del cartílago y tiene una gran variedad de manifestaciones articulares. La membrana sinovial hiperplásica interviene en el proceso invadiendo profundamente el cartílago articular y el resto de la articulación. En este proceso hay una gran variedad de mediadores, tanto inflamatorios como no inflamatorios, incluidas las citocinas proinflamatorias (interleucina [IL]-1β, TNF [tumor necrosis factor ‘factor de necrosis tumoral’] α, metaloproteinasas, células CD4+, linfocitos B, macrófagos y fibroblastos sinoviales), que contribuyen a la patogénesis de la AR.
Aunque la eficacia de los nuevos fármacos para tratar la AR está comprobada, ésta es variable. Mientras no haya ningún marcador molecular o clínico fiable y útil en respuesta al tratamiento, las concentraciones de varias citocinas u otros mediadores de la inflamación se pueden correlacionar con la eficacia de los tratamientos. La farmacogenética se centra en el estudio de los polimorfismos de aquellos genes que codifican para enzimas que metabolizan los fármacos, aunque en la actualidad también se centra en los polimorfismos de los transportadores de fármacos así como en las dianas de actuación de éstos1.
En esta revisión se refleja, por una parte, la influencia clínica de algunos polimorfismos presentes en genes que están relacionados con la AR y, por otra parte, los principios de la farmacogenética aplicada a distintos tratamientos, como los clásicos fármacos antirreumáticos modificadores de la enfermedad (FAME) y los nuevos agentes biológicos. En el futuro inminente, los estudios farmacogenéticos podrán ayudar a seleccionar los medicamentos y la dosis adecuada para cada sujeto.
Influencia clínica de los polimorfismos genéticos en la artritis reumatoideMuchas enfermedades son multifactoriales; en ellas, tanto el ambiente como los factores genéticos contribuyen a la etiología o a la gravedad clínica. La genética de muchas enfermedades multifactoriales es compleja, ya que se ven involucrados varios genes y además no se aplica el modelo de herencia mendeliano. La contribución genética a la susceptibilidad de la AR se ve reflejada tanto en grupos familiares como, sobre todo, en gemelos monocigóticos2. Varios autores sugieren que por lo menos 10 regiones genéticas distintas pueden estar relacionadas con la AR3. La variabilidad en la contribución de múltiples factores genéticos involucrados en la AR puede tener relación con la variabilidad que se refleja en las manifestaciones clínicas, que oscila entre una enfermedad leve y una enfermedad grave. Por otra parte, la variabilidad en la respuesta a los medicamentos es más acentuada entre la población que en un mismo donante o entre gemelos monocigóticos. Parte de esta diferencia se atribuye a factores genéticos4.
La mayoría de los genes implicados en la predisposición al desarrollo de la AR se localizan en los loci del HLA (human leukocyte antigen ‘antígeno de histocompatibilidad’) DR5. Otros genes candidatos son aquellos que codifican para diversas citocinas. Las citocinas son mediadores importantes en la inflamación y desempeñan un papel tanto en la patofisiología de la inflamación articular como en la destrucción que tiene lugar en la progresión de la AR6.
El complejo antígeno de histocompatibilidadEl MHC (major histocompatibility complex ‘complejo principal de histocompatibilidad’) es una región genética que se ha asociado constantemente a la AR. La contribución de esta región es de aproximadamente un 30% del efecto genético total7. La AR está asociada a alelos específicos de HLA-DRB1 que codifican para una secuencia conservada de aminoácidos (residuos 70-74 en la cadena DRβ1) conocida como epítopo compartido8. Esta secuencia se encuentra en el suelo de la hendidura para el antígeno (denominado peptide-binding groove ‘sitio de unión al péptido’). Los alelos que llevan esta secuencia son DRB1*0401, DRB1*0404, DRB1*0405, DRB1*0408, DRB1*0101, DRB1*0102 y DRB1*10019. La presencia y el número de copias de los alelos de HLA-DRB1 que codifican para el epítopo compartido se han asociado a la presencia de nódulos reumáticos, a un mayor y más rápido desgaste de la articulación, al síndrome de Felty, a vasculitis y en muchos casos a la necesidad de cirugía10. El genotipo DRB1*0401/DRB1*0404 está aparentemente asociado a enfermedades con un inicio prematuro, así como a un fenotipo más grave9.
También se han descrito secuencias de tipo microsatélite dentro de la región HLA; así, el transcrito 2 asociado al alelo HLA-B (BAT2) y el D6S273 son microsatélites de la región HLA de clase iii, mientras que el D62223 es un microsatélite de la región HLA de clase i11. En esta revisión se describirá cómo algunos de estos marcadores microsatélites están relacionados con la respuesta al tratamiento.
Genes de citocinas en la artritis reumatoideSi se considera el papel crítico de varias citocinas (como el TNF y la IL-1) en la patogénesis de la AR y se considera la heterogeneidad de la regulación genética de éstas, así como la presencia de estas moléculas en la articulación, es posible que los polimorfismos que regulan la producción de estas citocinas afecten el curso natural de la enfermedad12. Recientemente se ha identificado un gran número de polimorfismos con posibles fenotipos funcionales (tabla 1), mayoritariamente en la región promotora de varias citocinas, y se sospecha que son de gran importancia para mantener el equilibrio entre citocinas proinflamatorias y antiinflamatorias.
Polimorfismos genéticos en la artritis reumatoide
| Símbolo del gen | Posición del polimorfismo | Alelo | Posible efecto del polimorfismo | Bibliografía |
| TNF-α | +1304 | G | Puede contribuir a la susceptibilidad a AR. Posible desequilibrio de ligamiento | 22 |
| A | ||||
| +489 | G | Erosión articular más grave | 16 | |
| A | ||||
| −238 | G | Erosiones articulares más graves | 20 | |
| A | Erosiones articulares menos graves | |||
| −308 | G | Producción normal de TNF-α | 18,19 | |
| A | Regulación positiva de la producción de TNF-α | |||
| −857 | C | Puede contribuir a la susceptibilidad a AR. Alta producción de TNF-α | 23 | |
| T | ||||
| −863 | C | Puede contribuir a la susceptibilidad a AR. Alta producción de TNF-α | 21 | |
| A | ||||
| −1031 | T | Puede contribuir a la susceptibilidad a AR. Alta producción de TNF-α | 22,24 | |
| C | ||||
| TNFa6; b5; c1; d3; e3 | Incremento en la susceptibilidad a AR | 29 | ||
| TNFRSF1B | Codón 196 | T | Aumento más efectivo en la producción de IL-6 | 26 |
| G | ||||
| IL-1 | IL-1α −889 | C | Producción alterada de IL-1α | 42 |
| T | ||||
| IL-1α +4845 (exón 5) | G | Producción alterada de IL-1α. Incremento en la susceptibilidad a AR | 34,38,41 | |
| T | ||||
| IL-1β −511 | C | Producción alterada de IL-1α | 40 | |
| T | ||||
| IL-1β +3953 (exón 5) | C | Producción alterada de IL-1RA. Destrucción grave de la articulación | 39,40,41 | |
| T | ||||
| IL-1RA +2018 (exón 2) | C | Posible efecto proinflamatorio | 43 | |
| T | ||||
| IL-6 | −174 | G | Disminuye la producción de IL-6 | 49 |
| C | ||||
| −622 | G | |||
| A | Disminuye la producción de IL-6 | 48 | ||
| IL-10 | −1082 | G | Regulación positiva de la producción de IL-10 en linfocitos | 12 |
| A | Concentraciones bajas de IL-10. Se asocia a AR en mujeres | 52,54 | ||
| −819 | T | Concentraciones bajas de IL-10. Manifestaciones autoinmunes | 52,53 | |
| C | ||||
| −592 | A | Concentraciones de IL-10. Manifestaciones autoinmunes | 53,52 | |
| C | ||||
| HLA | Alelos específicos del epítopo compartido (HLA-DR) | Puede contribuir a la susceptibilidad y gravedad de la AR | 8–10 |
A: adenina;AR: artritis reumatoide; C: ; G: guanina; HLA: antígeno de histocompatibilidad; IL-1: interleucina-1; IL-6: interleucina-6; IL-1RA: antagonista del receptor de IL-1; T: ; TNF-α: factor de necrosis tumoral alfa; TNFRSF1B: receptor de tipo 2 del TNF-α.
Una de las moléculas que desempeña un papel importante en la patogénesis de la AR es la citocina proinflamatoria TNF. Esta molécula pertenece a una familia de proteínas involucradas en la regulación del sistema inmune y en la programación de la muerte celular. Las concentraciones de TNF en sujetos con AR están elevadas crónicamente en sangre y más específicamente en las articulaciones13. Dos receptores intervienen en las funciones de esta molécula: TNFRSF1A y TNFRSF1B, que están presentes como monómeros tanto en la superficie celular como en las formas solubles14. Se sabe que el TNF está involucrado en la estimulación de la producción de citocinas (incrementa la expresión de moléculas adherentes) y en la activación de neutrófilos.
El TNF es también un coestimulador de la activación de células T y de la producción de anticuerpos por células B15. Además, contribuye a la regulación de la homeostasis y desempeña un papel importante en la inflamación. Un 60% de la variación en la producción de TNF está determinada genéticamente, lo que indica una influencia genética sobre la producción de citocinas16. Todas estas características, además de su localización en el cromosoma 6 en la región MHC clase iii entre los genes HLA-B y HLA-DR17, permiten especular con la existencia de polimorfismos funcionales en este gen. Como consecuencia de todo lo descrito, se ha considerado al gen TNF como un gen candidato a estar asociado a la enfermedad.
Dentro del gen TNF se ha descrito la presencia, mayoritariamente en la región promotora, de SNP (single nucleotide polymorphism ‘polimorfismos mononucleotídicos’) (fig. 1). El primer polimorfismo identificado fue una transición entre guanina (G) y adenina (A) en la posición −308. El alelo A, poco común, tiene una fuerte asociación al haplotipo HLA-A1-B8-DR3-DQ218 y también se asocia a enfermedades autoinmunes y a fenotipos que ocasionan una mayor producción de TNF19. Este alelo puede facilitar la desregulación de la red de citocinas y originar la AR16.
 alfa, donde se reflejan los polimorfismos de una sola base más relevantes. Las flechas horizontales indican la orientación transcripcional de los genes del TNF y de la linfotoxina. Las regiones de los exones 1 y 4 resaltadas en diagonal indican la región no traducida.")
Representación esquemática del gen del factor de necrosis tumoral (TNF) alfa, donde se reflejan los polimorfismos de una sola base más relevantes. Las flechas horizontales indican la orientación transcripcional de los genes del TNF y de la linfotoxina. Las regiones de los exones 1 y 4 resaltadas en diagonal indican la región no traducida.
Los estudios del polimorfismo situado en la posición −238 de la región promotora del TNF mostraron una mayor presencia del alelo G frente al alelo A. De los 3 posibles genotipos, GG y GA son los más comunes; el primero de éstos es el que parece estar asociado a erosiones articulares más graves, mientras que los sujetos que portan el genotipo GA presentan un deterioro más lento20. Otros estudios mostraron una asociación similar en la posición +489: los individuos con el genotipo GG en esta posición reflejaban una erosión más grave en el desarrollo de la enfermedad16.
Los polimorfismos descritos anteriormente, así como otros presentes en este gen, tales como −1031 T/C, −863 C/A, −857 C/T o +1304 G/A, pueden contribuir a la susceptibilidad de la AR debido a un incremento en la producción de TNF-α21–24, y pueden participar en varios haplotipos debido tanto al gran número de polimorfismos potencialmente relevantes como a los patrones complejos de desequilibrio de ligamiento que tienen lugar en la región del MHC25.
Se ha descrito un SNP en el exón 6 del receptor de tipo 2 del TNF-α (TNFRSF1B), que consiste en una sustitución de una única base en el codón 196 (T a G, de ATG a AGG), que implica un cambio aminoacídico no conservado (de metionina a arginina). El alelo 196G parece ser más efectivo en la producción de IL-6 que el alelo 196T. El alelo 196G puede afectar también a los receptores de membrana26.
En el locus del TNF, al igual que en el caso anterior, también se han descrito secuencias de ácido desoxirribonucleico microsatélite. Las repeticiones consisten en secuencias de bases A y T y se localizan en regiones no codificantes. Estas secuencias sirven como marcadores genéticos cuando se encuentran en desequilibro de ligamiento con un polimorfismo funcional en las proximidades del gen27. El locus del TNF tiene 5 microsatélites (de TNFa a TNFe) que se basan en el número de secuencias repetidas28. Estudios in vitro indican que TNFd y TNFa2 están asociados a concentraciones altas de TNF-α, mientras que TNFa6 está asociado a concentraciones bajas28. Algunos haplotipos microsatélite, como TNFa6; TNFb5; TNFc1; TNFd3, y TNFe3, se han asociado al aumento de la susceptibilidad a presentar AR29.
Interleucina-1La IL-1 es otra citocina que contribuye a la destrucción crónica que tiene lugar en la AR. Se acepta que la artritis puede inducirse en ratones mediante una inyección local de citocinas recombinantes (TNF o IL-1) en la articulación de la rodilla30. La actividad biológica de la IL-1 depende del equilibrio entre 2 citocinas proinflamatorias (IL-1α y IL-1β) y una proteína antiinflamatoria (el antagonista del receptor de IL-1 [IL-1RA]). El IL-1RA bloquea la unión de IL-1α y IL-1β a su receptor y regula la activación de estas 2 citocinas. La IL-1 es importante ya que induce la supresión de la síntesis de la matriz llevada a cabo por los condrocitos y la liberación de agrecanasas, enzimas causantes de la pérdida de proteoglucanos30.
Los genes que codifican estas 3 proteínas (IL-1α, IL-1β y IL-1RA) están localizados en una región de 430kb en el cromosoma 231. En cada uno de estos genes hay SNP y otros tipos de alteraciones que originan la existencia de haplotipos comunes en la población, dado el elevado desequilibrio de ligamiento que tiene lugar en esta región32. Entre los polimorfismos de interés se encuentran: a) SNP bialélicos en el gen IL-1 α localizados en la posición −889 C/T33 y en el exón 5 en la posición +4845 G/T34; b) en el gen IL-1β en la posición −511 C/T35 y en el exón 5 en la posición +3953 C/T36; c) en el gen IL-1RA en la posición +2018 C/T en el exón 237, y d) un sitio polimorfito pentaalélico en el intrón 2 que contiene un VNTR (variable number of tandem repeats ‘número variable de repeticiones en tándem’) de una secuencia de 86 bp.
Algunos estudios han reflejado la asociación entre la presencia de los alelos menos prevalentes en los genes IL-1α (+4845) o IL-1β (+3953) y un incremento tanto en la susceptibilidad a la AR38 como en la destrucción de la articulación39. Otros estudios han relacionado algunos de los polimorfismos descritos anteriormente, IL-1α (−889), IL-1 α(+4845), IL-1β (+3953) o el VNTR del intrón 2 de IL-1RA, con una producción alterada de IL-140–42. También se ha descrito que el genotipo en la IL-1β puede influir en las concentraciones de IL-1RA40. Por otra parte, el polimorfismo IL-1RA +2018 C/T parece tener un efecto proinflamatorio43.
Interleucina-6La IL-6 es otra citocina pleiotrópica con un gran rango de actividades biológicas, incluidas la regulación de la respuesta inmune, la inflamación, la hematopoyesis y el metabolismo del hueso44. La sobreproducción de IL-6 parece tener un papel en la patogénesis de la AR. En algunos trabajos se ha descrito que las concentraciones de IL-6 en suero tienen una correlación con la actividad de la enfermedad y con daños en las articulaciones detectados en radiografías45. Sin embargo, otros autores proponen que la IL-6 actúa como un mediador antiinflamatorio46. Se ha observado que la IL-6 aumenta las concentraciones de IL-1RA y de receptores solubles de TNF en la circulación, y ambos aspectos podrían provocar un efecto antiinflamatorio, ya que suprimiría la acción de IL-1 y de TNF47.
Se han descrito polimorfismos en la región promotora de la IL-6, entre los que se encuentran una transversión de G/C en la posición −174 y una transición de G/A en la posición −622; ambos se encuentran en completo desequilibrio de ligamiento48. Se demostró que el polimorfismo de la posición −174 afecta las concentraciones de IL-6 y se ha asociado a la artritis idiopática juvenil sistémica49. Sin embargo, los últimos datos parecen descartar la importancia del papel de estos polimorfismos en la susceptibilidad a la AR48,50.
Interleucina-10Otra citocina que media en la regulación de la respuesta inflamatoria es la IL-10, que actúa como regulador negativo de TNF-α y de otras citocinas proinflamatorias51. Hay distintos polimorfismos en la IL-10 (su gen está localizado en el cromosoma 1) que pueden afectar a las concentraciones de las citocinas producidas. Así, las alteraciones puntuales en las posiciones −1082 G/A, −819 T/C y −592 A/C pueden dar como resultado el haplotipo ACC que se asocia a valores bajos de expresión de IL-1052. Estas variaciones también se correlacionan con manifestaciones autoinmunes53, en particular el genotipo -1082AA se asocia al desarrollo de la AR en mujeres54. Por el contrario, el genotipo -1082GG de esta citocina se asocia a una regulación positiva en la producción de IL-10 por parte de los linfocitos12.
Farmacogenómica de fármacos antirreumáticosFármacos antirreumáticos modificadores de la enfermedad en la artritis reumatoideEstos fármacos tienen la capacidad de reducir o de prevenir los daños en las articulaciones y preservan la integridad y la función de éstas al afectar al sistema inmune. Sin embargo, las consecuencias del tratamiento con estos fármacos en sujetos diagnosticados de AR son variables e imprevisibles. Una posible causa para explicar las diferencias tanto en la eficacia como en la aparición de reacciones adversas pueden ser las variaciones genéticas que presentan los individuos al metabolizar estos fármacos.
Los FAME que tienen un potencial uso como fármacos personalizados en función del perfil genético del sujeto con AR son el metotrexato (MTX), la sulfasalazina (SSZ) y la azatioprina (AZT).
MetotrexatoEste fármaco es el más utilizado para el tratamiento de la AR, y su principal efecto farmacológico parece ser el antagonismo del folato. El MTX entra en la célula a través del RFC (reduced folate carrier, ‘transportador de folato reducido’) 1 y se convierte intracelularmente en poliglutamatos de MTX, lo que favorece la retención intracelular de MTX al promover la inhibición de la síntesis de purinas así como la formación de adenosina, un potente agente antiinflamatorio.
El MTX inhibe directamente varias enzimas, como la dihidrofolato reductasa, la 5-aminoimidazol-4-carboxamida ribonucleótido (AICAR), la transformilasa (ATIC) o la timidilato sintasa (TYMS). El MTX inhibe directamente a otras enzimas, como la metilenotetrahidrofolato reductasa (MTHFR), pero su grado de expresión puede contribuir a incrementar los efectos del MTX.
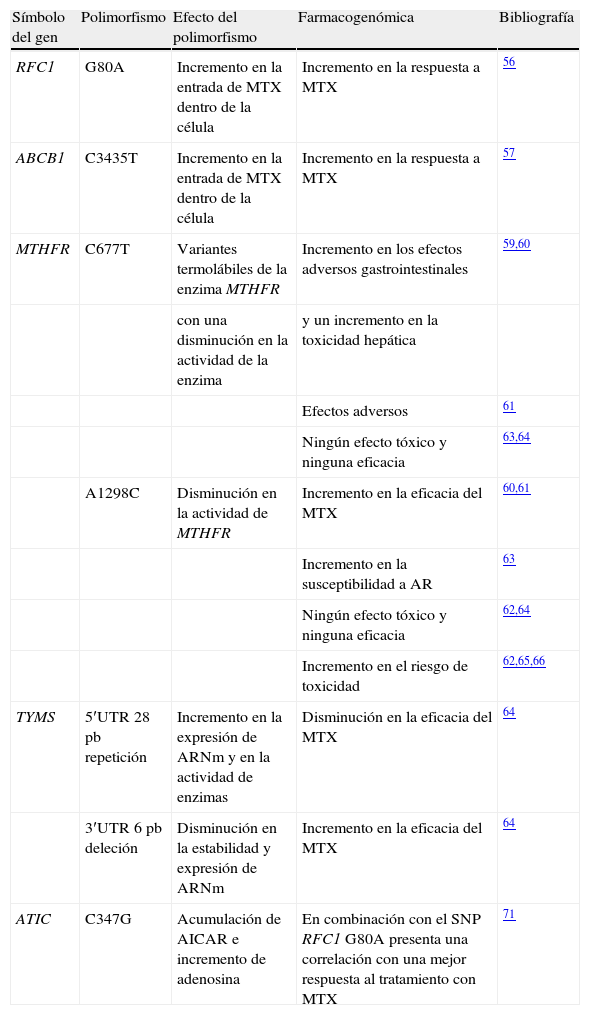
Una revisión extensa de la farmacogenética del MTX55 muestra la existencia de varios polimorfismos genéticos relacionados con los transportadores de MTX a través de la membrana celular y con las enzimas influyentes en su ruta metabólica (tabla 2). Entre los polimorfismos que influyen en el transporte del MTX a través de la membrana celular destacan los polimorfismos G80A en el RFC1 y el C3435T en el gen ABCB1, que codifica para un transportador de membrana (glucoproteína P) que es el que realmente está implicado en la biodisponibilidad y disposición de distintos fármacos. Variaciones genéticas en estos transportadores pueden afectar la respuesta frente al MTX en sujetos con AR, ya que ambos incrementan la entrada del fármaco en la célula. Aquellos individuos que presentan el genotipo RFC 80A/A tienen una mejor respuesta frente a los sujetos portadores del alelo salvaje (80G/G)56. Los sujetos con los genotipos ABCB1 3435C/C y 3435 C/T tienen un mayor riesgo de presentar AR, comparados con sujetos poseedores del genotipo 3435T/T, ya que tienen una mejor respuesta al tratamiento con MTX57.
Datos de farmacogenética relacionados con la eficacia o la toxicidad por metotrexato en la artritis reumatoide
| Símbolo del gen | Polimorfismo | Efecto del polimorfismo | Farmacogenómica | Bibliografía |
| RFC1 | G80A | Incremento en la entrada de MTX dentro de la célula | Incremento en la respuesta a MTX | 56 |
| ABCB1 | C3435T | Incremento en la entrada de MTX dentro de la célula | Incremento en la respuesta a MTX | 57 |
| MTHFR | C677T | Variantes termolábiles de la enzima MTHFR | Incremento en los efectos adversos gastrointestinales | 59,60 |
| con una disminución en la actividad de la enzima | y un incremento en la toxicidad hepática | |||
| Efectos adversos | 61 | |||
| Ningún efecto tóxico y ninguna eficacia | 63,64 | |||
| A1298C | Disminución en la actividad de MTHFR | Incremento en la eficacia del MTX | 60,61 | |
| Incremento en la susceptibilidad a AR | 63 | |||
| Ningún efecto tóxico y ninguna eficacia | 62,64 | |||
| Incremento en el riesgo de toxicidad | 62,65,66 | |||
| TYMS | 5′UTR 28 pb repetición | Incremento en la expresión de ARNm y en la actividad de enzimas | Disminución en la eficacia del MTX | 64 |
| 3′UTR 6 pb deleción | Disminución en la estabilidad y expresión de ARNm | Incremento en la eficacia del MTX | 64 | |
| ATIC | C347G | Acumulación de AICAR e incremento de adenosina | En combinación con el SNP RFC1 G80A presenta una correlación con una mejor respuesta al tratamiento con MTX | 71 |
ABCB1: adenosintrifosfato-binding cassette B1; AICAR: 5-aminoimidazol-4-carboxamida ribonucleótido; AR: artritis reumatoide; ARNm: ácido ribonucleico mensajero; ATIC: aminoimidazol carboxamida ribonucleótido transformilasa; MTHFR: metilenotetrahidrofolato reductasa; MTX: metotrexato; RFC1: transportador del folato reducido; SNP: polimorfismo mononucleotídico; TYMS: timidilato sintasa; UTR: región no traducida.
Entre los polimorfismos que influyen en las enzimas metabólicas implicadas en la ruta celular de este fármaco destacan 2 SNP localizados en el gen que codifica para la enzima MTHFR. Esta enzima es muy importante para la regeneración de folato reducido. El polimorfismo C677T en este gen da como resultado una variante termolábil con detrimento de la actividad enzimática58. Hay un gran rango de efectos clínicos asociados a estos polimorfismos, como incremento de efectos adversos gastrointestinales59, aumento en la toxicidad hepática60 y diversos efectos adversos61. Además, los últimos estudios muestran que los portadores del genotipo MTHFR 677TT responden menos al MTX en comparación con otros genotipos62; sin embargo, otros autores no encontraron ningún efecto sobre la toxicidad ni sobre la eficacia63,64. El polimorfismo A1298C confiere una actividad disminuida del MTHFR y también muestra discrepancias en sus efectos clínicos. Así, algunos estudios proponen la existencia de una eficacia incrementada de MTX60,61, una mayor susceptibilidad para presentar AR63 y un incremento en la toxicidad62,65,66; sin embargo, en otro estudio no se detectó ningún efecto sobre la eficacia y la toxicidad64.
Los genes que codifican para TYMS y ATIC también están relacionados con la ruta celular del MTX, ya que son dianas de éste. La TYMS es una enzima clave en la síntesis de novo de timidilato y convierte deoxiuridina monofosfato en deoxitimidina monofosfato. Poliglutamatos de MTX inhiben a esta enzima. En la región 5′-UTR (untranslated region ‘región no traducida’) del gen TYMS se identificó una repetición en tándem polimórfica, con un VNTR de 28pb67; cuanto mayor sea el número de elementos repetidos, mayor será la expresión de ácido ribonucleico mensajero (ARNm) y mayor será la actividad enzimática68, de manera que disminuye la eficacia del MTX64. En este gen se ha descrito otro polimorfismo que consiste en una deleción de 6-pb (TTAAAG) en la posición 1496 de la región 3′-UTR69, que puede asociarse a una disminución en la estabilidad y expresión del ARNm70 de este gen, de manera que se incrementa la eficacia del MTX64.
La ATIC convierte aminoimidazol carboxamida ribonucleotido en 10-formil AICAR. El MTX inhibe directamente a la ATIC, con lo que se origina una acumulación de AICAR y de adenosina, una purina antiinflamatoria. Un estudio previo determinó que la homocigosidad para el polimorfismo C347G en ATIC y la presencia del SNP G80A en RFC1 pueden estar relacionadas con una mejor respuesta al MTX71.
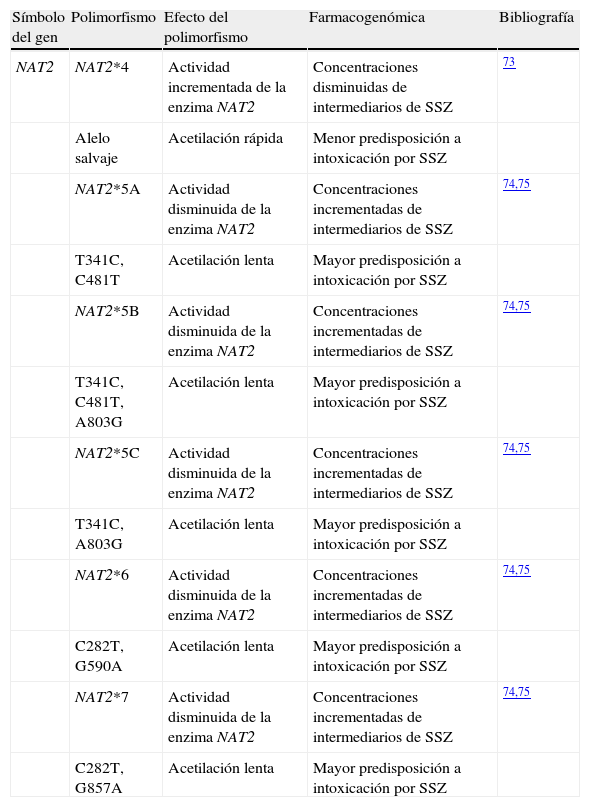
SulfasalazinaLa SSZ es otro fármaco englobado dentro de los FAME con un uso común para el tratamiento de la AR. Sin embargo, su uso está limitado debido a sus efectos adversos72. Después de la ingestión oral, bacterias intestinales en 5-amino ácido salicílico y sulfapiridina escinden la SSZ, y este último compuesto se metaboliza en el hígado por acetilación. El gen NAT2 localizado en el cromosoma 8p22 codifica la enzima involucrada en la acetilación de sulfapiridina y puede ser polimórfico. Los polimorfismos génicos en NAT2(tabla 3) influyen en el estado de acetilación lento frente al estado de acetilación rápido en un individuo. Las acetilaciones lentas son más propensas a toxicidad por SSZ, comparadas con las rápidas73.
Datos de farmacogenética relacionados con la eficacia o la toxicidad por sulfasalazina en la artritis reumatoide
| Símbolo del gen | Polimorfismo | Efecto del polimorfismo | Farmacogenómica | Bibliografía |
| NAT2 | NAT2*4 | Actividad incrementada de la enzima NAT2 | Concentraciones disminuidas de intermediarios de SSZ | 73 |
| Alelo salvaje | Acetilación rápida | Menor predisposición a intoxicación por SSZ | ||
| NAT2*5A | Actividad disminuida de la enzima NAT2 | Concentraciones incrementadas de intermediarios de SSZ | 74,75 | |
| T341C, C481T | Acetilación lenta | Mayor predisposición a intoxicación por SSZ | ||
| NAT2*5B | Actividad disminuida de la enzima NAT2 | Concentraciones incrementadas de intermediarios de SSZ | 74,75 | |
| T341C, C481T, A803G | Acetilación lenta | Mayor predisposición a intoxicación por SSZ | ||
| NAT2*5C | Actividad disminuida de la enzima NAT2 | Concentraciones incrementadas de intermediarios de SSZ | 74,75 | |
| T341C, A803G | Acetilación lenta | Mayor predisposición a intoxicación por SSZ | ||
| NAT2*6 | Actividad disminuida de la enzima NAT2 | Concentraciones incrementadas de intermediarios de SSZ | 74,75 | |
| C282T, G590A | Acetilación lenta | Mayor predisposición a intoxicación por SSZ | ||
| NAT2*7 | Actividad disminuida de la enzima NAT2 | Concentraciones incrementadas de intermediarios de SSZ | 74,75 | |
| C282T, G857A | Acetilación lenta | Mayor predisposición a intoxicación por SSZ |
NAT2: N-acetiltransferasa 2; SSZ: sulfasalazina.
El alelo salvaje NAT2*4 codifica para el mecanismo de acetilación rápida, mientras que sus variantes (NAT2*5A, NAT2*5B, NAT2*5C, NAT2*6 y NAT2*7), que difieren en combinaciones de varios SNP localizados en el exón 2 del gen, codifican para el mecanismo de acetilación lenta, lo que se traduce en un detrimento de la actividad de la enzima NAT2. A causa de las acetilaciones lentas, estas variantes están asociadas a concentraciones incrementadas de intermediarios de SSZ74,75.
El estado de acetilación de un individuo, que parece verse influenciado por los polimorfismos del gen NAT2, puede ser importante a la hora de determinar el riesgo de toxicidad frente a SSZ. Por tanto, sería útil en la práctica clínica desarrollar estudios para identificar el genotipo NAT2 en sujetos antes de iniciar el tratamiento con SSZ para prevenir de la toxicidad de este fármaco76.
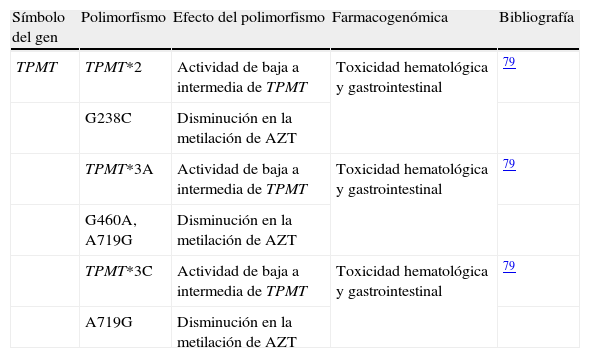
AzatioprinaLa AZT es un medicamento utilizado en el tratamiento de diferentes tipos de cáncer, en enfermedades reumáticas y en el rechazo de órganos trasplantados. Pese a esto, la AZT no se emplea con mucha frecuencia en el tratamiento de la AR debido, entre otras causas, al desarrollo de otros FAME. La tiopurina metiltransferasa (TPMT) es una de las enzimas involucradas en el metabolismo de este fármaco. Diferentes estudios poblacionales han permitido establecer que la actividad de la TPMT en eritrocitos es trimodal: aproximadamente un 90% de la población tiene una actividad alta, un 10% tiene una actividad intermedia y únicamente el 0,3% tiene muy poca o ninguna actividad77.
Hay 3 variantes alélicas del gen TPMT: TPMT*2 (G238C), TPMT*3A (G460A y A719G) y TPMT*3C (A719G) (tabla 4) presentes en el 60 al 95% de la población que presenta una actividad baja o intermedia de TPMT78. Clínicamente, estos polimorfismos están asociados a toxicidad hematológica y, en algunos casos, gastrointestinal79. El genotipo del gen TPMT puede ser útil a la hora de predecir la toxicidad a AZT.
Datos de farmacogenética relacionados con la toxicidad de la azatioprina en la artritis reumatoide
| Símbolo del gen | Polimorfismo | Efecto del polimorfismo | Farmacogenómica | Bibliografía |
| TPMT | TPMT*2 | Actividad de baja a intermedia de TPMT | Toxicidad hematológica y gastrointestinal | 79 |
| G238C | Disminución en la metilación de AZT | |||
| TPMT*3A | Actividad de baja a intermedia de TPMT | Toxicidad hematológica y gastrointestinal | 79 | |
| G460A, A719G | Disminución en la metilación de AZT | |||
| TPMT*3C | Actividad de baja a intermedia de TPMT | Toxicidad hematológica y gastrointestinal | 79 | |
| A719G | Disminución en la metilación de AZT |
AZT: azatioprina; TPMT: tiopurina metiltransferasa.
La introducción de los agentes biológicos ha alterado notablemente el tratamiento de la AR; estos agentes no sólo reducen los síntomas y los signos de la enfermedad, sino que también retrasan la progresión radiológica de ésta80. Sin embargo, estos tratamientos son sustancialmente más caros que los FAME tradicionales y además no son efectivos en todos los sujetos81. Hay estudios que reflejan que entre un 25 y un 30% de los sujetos con AR no responde a estos tratamientos82. La identificación temprana de sujetos que respondan positivamente a estos fármacos puede ser de gran ayuda para establecer un tratamiento efectivo con estas moléculas83.
Diversos estudios de los procesos inflamatorios mediados por TNF e IL-1 han conducido al desarrollo de agentes que bloquean las citocinas para el tratamiento de la AR. Tres bloqueadores de TNF están aprobados por la FDA para el tratamiento de la AR: etanercept, infliximab y adalimumab. Estos bloqueadores derivan de un receptor recombinante de TNF (TNFRSF1B) para etanercept o de un anticuerpo monoclonal anti-TNF-α en el caso del infliximab y del adalimumab. El mecanismo molecular de estos distintos bloqueadores de TNF se basa en el mismo principio, que consiste en impedir la unión del TNF a los receptores celulares de superficie para el TNF, de este modo se bloquea la vía de transducción de señales inducidas o reguladas por el TNF. Aunque el tratamiento neutralizante anti-TNF-α puede ser muy efectivo al reducir los síntomas y las indicaciones de AR, no todos los sujetos muestran el mismo grado de respuesta en la progresión de la enfermedad84. Se ha propuesto que la variabilidad en las regiones promotora y codificante del gen TNF-α pueden modular la magnitud de la respuesta secretora de esta citocina85.
El cuarto agente biológico aprobado por la FDA para el tratamiento de la AR es la anakinra, una forma recombinante del IL-1RA; su mecanismo molecular ya se ha explicado anteriormente.
Todos los fármacos que tienen el potencial para usarse en un «tratamiento hecho a la medida» dirigido a sujetos con AR, comparten problemas relacionados con la efectividad y la toxicidad. En respuesta a la toxicidad de estos fármacos, se ha descrito el riesgo de presentar linfoma, pero hay que tener en cuenta que los sujetos con AR grave presentan de por sí un riesgo 2 veces mayor de tener un linfoma. Sólo algunos de estos linfomas están relacionados con la presencia del virus de Epstein-Barr. Esto puede, a su vez, estar relacionado con la presencia elevada de este tipo de virus en sujetos con AR, lo que refleja una ligera discapacidad de la inmunidad antivírica en estos sujetos86.
EtanerceptEl etanercept es una proteína de fusión dimérica, que incluye el receptor p75 del TNF humano unido a un dominio del fragmento c de la inmunoglobulina G1 (IgG1) (fig. 2a). Este medicamento se fabrica exclusivamente con secuencias de aminoácidos humanos y consta de 934 aminoácidos.
 Bloqueador del factor de necrosis tumoral alfa (TNF-α), etanercept. b) Bloqueador del TNF-α, infliximab. c) Bloqueador del TNF-α, adalimumab. d) Tratamiento con el bloqueador de interleucina-1 (IL-1). Anakinra, una forma recombinante del antagonista del receptor de IL-1 humano IL-1 (IL-1RA).")
Mecanismos moleculares. a) Bloqueador del factor de necrosis tumoral alfa (TNF-α), etanercept. b) Bloqueador del TNF-α, infliximab. c) Bloqueador del TNF-α, adalimumab. d) Tratamiento con el bloqueador de interleucina-1 (IL-1). Anakinra, una forma recombinante del antagonista del receptor de IL-1 humano IL-1 (IL-1RA).
La eficacia del etanercept para el tratamiento de la AR se ha demostrado tanto en monoterapia como en combinación con MTX. La progresión de la degeneración de la articulación disminuye significativamente cuando este fármaco se suministra durante más de 24 meses. Además, el tratamiento con etanercept es significativamente mejor que la monoterapia con MTX.
Sin embargo, se han descrito infecciones oportunistas, tuberculosis, insuficiencia cardíaca y linfoma en algunos sujetos que recibieron tratamiento con este fármaco. El riesgo de presentar alguna de éstas se ve incrementado si al mismo tiempo de seguir este tratamiento el sujeto ingiere corticoides u otros agentes inmunosupresores. Además, también se ha descrito el riesgo de presentar enfermedades desmielinizantes87.
InfliximabEl infliximab es un anticuerpo monoclonal que se une al TNF-α y neutraliza su actividad. Es una quimera de ratón con humano en la que se unen las regiones variables del anticuerpo del ratón con la región constante de la IgG1 de humanos (fig. 2b). Este fármaco es el primer bloqueador de TNF usado en el tratamiento de la AR y muestra la importancia que desempeña el TNF-α en esta enfermedad.
El tratamiento con infliximab ha proporcionado a los sujetos una mejora en la calidad de vida, la prevención de daños estructurales en la articulación y posiblemente la reparación del hueso. Este medicamento también se ha usado con éxito, solo o combinado con MTX, para el tratamiento de otras enfermedades, como la enfermedad de Crohn, la espondilitis anquilosante o la artritis psoriásica.
Por el contrario, el infliximab presenta los mismos problemas que el etanercept: reacciones locales en el punto de inyección, infecciones oportunistas, tuberculosis y un mayor riesgo de desarrollo de linfoma88. Del mismo modo, se han descrito procesos de desmielinizacion, insuficiencia cardíaca y enfermedades autoinmunes.
AdalimumabEl adalimumab es un anticuerpo monoclonal humano IgG1. Su mecanismo molecular es parecido al del infliximab: se une tanto al TNF circulante como al TNF de las superficies celulares bloqueando la interacción del TNF-α con sus receptores p55 y p75 localizados en la superficie de las células (fig. 2c). El adalimumab modula la respuesta biológica inducida por el TNF y reduce las concentraciones de IL-6 y metaloproteinasas de la matriz (MPM) 1 y MPM-389.
El adalimumab se utiliza en monoterapia o combinado con MTX. Un beneficio destacado de este fármaco es la inhibición de la progresión de los daños estructurales de la articulación a largo plazo en sujetos con AR que no han respondido satisfactoriamente a otros FAME.
Los efectos tóxicos asociados al uso de adalimumab incluyen los mismos problemas descritos para los 2 tratamientos anteriores: infecciones oportunistas, tuberculosis, procesos de desmielinización, enfermedades autoinmunes e insuficiencia cardíaca.
AnakinraLa anakinra es una forma recombinante de IL-1RA humano que actúa como un antagonista de la actividad biológica de la IL-1 por inhibición competitiva, y se une al receptor de la membrana celular de la IL-1 y bloquea la señalización celular (fig. 2d). Su eficacia en sujetos con AR se ha investigado en monoterapia y también combinado con MTX, etanercept y otros FAME90–92. Asimismo, puesto que la IL-1 desempeña un papel importante en el desarrollo de la enfermedad de Still, este fármaco ha sido empleado con éxito para el tratamiento de esa enfermedad93,94. El tratamiento combinado con etanercept no ha mostrado ventajas clínicas y actualmente está contraindicado, ya que aumenta el desarrollo de infecciones oportunistas.
La inhibición de los daños estructurales es un beneficio importante de este fármaco, mientras que la reacción en el punto de inyección es su mayor desventaja; además, se han descrito neumonías e infecciones cutáneas92.
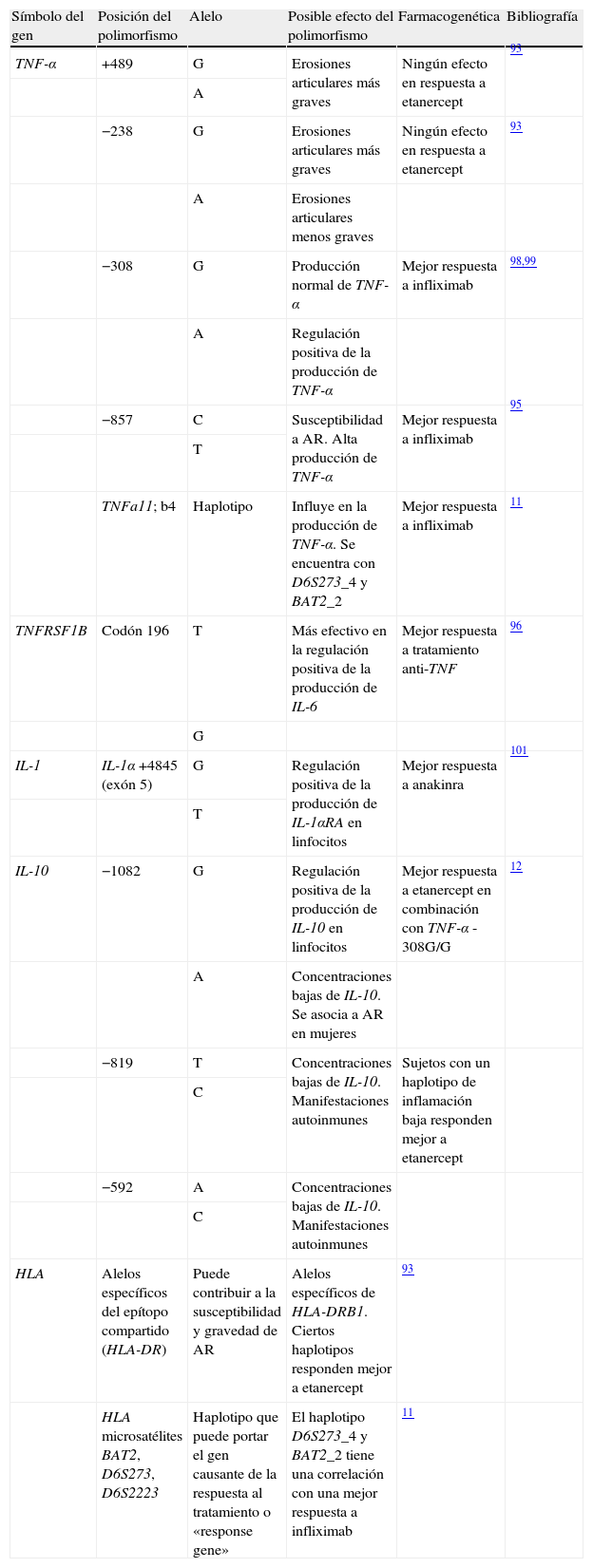
Farmacogenómica de los agentes biológicosVarios estudios han correlacionado la respuesta a los agentes biológicos con alguno de los polimorfismos genéticos descritos en esta revisión (tabla 5). Así, diversos haplotipos que incluyen tanto la región HLA-DRB1 como la región correspondiente al gen del TNF influyen en la respuesta a etanercept en sujetos caucásicos95. En este mismo estudio se pone de manifiesto que aquellos sujetos que presentan 2 copias de alelos HLA-DRB1 del epítopo compartido obtienen una mejor respuesta al etanercept al compararlos con los sujetos que tenían una o ninguna copia de aquel alelo. Dado el gran número de genes localizados en la región HLA que influyen tanto en el funcionamiento como en la regulación del sistema inmune y la existencia de desequilibrio de ligamiento que tiene lugar en esta región son numerosos los genes que pueden influir en la respuesta al tratamiento96.
Datos de farmacogenética relacionados con la eficacia o la toxicidad de los agentes biológicos en la artritis reumatoide
| Símbolo del gen | Posición del polimorfismo | Alelo | Posible efecto del polimorfismo | Farmacogenética | Bibliografía |
| TNF-α | +489 | G | Erosiones articulares más graves | Ningún efecto en respuesta a etanercept | 93 |
| A | |||||
| −238 | G | Erosiones articulares más graves | Ningún efecto en respuesta a etanercept | 93 | |
| A | Erosiones articulares menos graves | ||||
| −308 | G | Producción normal de TNF-α | Mejor respuesta a infliximab | 98,99 | |
| A | Regulación positiva de la producción de TNF-α | ||||
| −857 | C | Susceptibilidad a AR. Alta producción de TNF-α | Mejor respuesta a infliximab | 95 | |
| T | |||||
| TNFa11; b4 | Haplotipo | Influye en la producción de TNF-α. Se encuentra con D6S273_4 y BAT2_2 | Mejor respuesta a infliximab | 11 | |
| TNFRSF1B | Codón 196 | T | Más efectivo en la regulación positiva de la producción de IL-6 | Mejor respuesta a tratamiento anti-TNF | 96 |
| G | |||||
| IL-1 | IL-1α +4845 (exón 5) | G | Regulación positiva de la producción de IL-1αRA en linfocitos | Mejor respuesta a anakinra | 101 |
| T | |||||
| IL-10 | −1082 | G | Regulación positiva de la producción de IL-10 en linfocitos | Mejor respuesta a etanercept en combinación con TNF-α -308G/G | 12 |
| A | Concentraciones bajas de IL-10. Se asocia a AR en mujeres | ||||
| −819 | T | Concentraciones bajas de IL-10. Manifestaciones autoinmunes | Sujetos con un haplotipo de inflamación baja responden mejor a etanercept | ||
| C | |||||
| −592 | A | Concentraciones bajas de IL-10. Manifestaciones autoinmunes | |||
| C | |||||
| HLA | Alelos específicos del epítopo compartido (HLA-DR) | Puede contribuir a la susceptibilidad y gravedad de AR | Alelos específicos de HLA-DRB1. Ciertos haplotipos responden mejor a etanercept | 93 | |
| HLA microsatélites BAT2, D6S273, D6S2223 | Haplotipo que puede portar el gen causante de la respuesta al tratamiento o «response gene» | El haplotipo D6S273_4 y BAT2_2 tiene una correlación con una mejor respuesta a infliximab | 11 |
A: adenina; AR: artritis reumatoide; C: ; G: guanina; HLA: antígeno de histocompatibilidad; IL-1: interleucina-1, IL-10: interleucina-10; T: ; TNF-α: factor de necrosis tumoral alfa; TNFRSF1B: receptor de tipo 2 del TNF-α.
En un extenso estudio, 78 sujetos tratados con infliximab fueron genotipados para los alelos HLA-DRB1, HLA-DQA1 y HLA-DQB1, para un polimorfismo repetitivo de trinucleótidos dentro del gen MICA, así como para microsatélites del gen del TNF desde el a hasta el e, y para microsatélites presentes en otras regiones del complejo HLA, como el D6S273, BAT2 (HLA clase iii) y el D6S2223 (HLA clase i). Al analizar todos los haplotipos, los autores de este estudio concluyeron que el par D6S273_4 y BAT2_2 es el más significativo en sujetos que responden al tratamiento. De la misma manera, la frecuencia del haplotipo TNFa11; b4, un marcador que normalmente se encuentra en D6S273_4 y BAT2_2, también se vio aumentado, mientras que la del alelo D6S273_3 se vio disminuida en sujetos que respondieron al tratamiento11. Estos resultados permitieron a los autores especular que estos marcadores pueden localizarse en el mismo haplotipo que incluya al hasta ahora desconocido «gen de respuesta».
Otros estudios han puesto de manifiesto una correlación entre los polimorfismos TNF-α -308G12 y TNF-α -857T97 y una buena respuesta a etanercept. Otros autores analizaron la respuesta a etanercept o infliximab en sujetos con AR grave, caracterizada por una respuesta negativa a MTX, en combinación con otros FAME. Los sujetos con el genotipo TNFRSFB 196TT tratados con tratamiento anti-TNF mostraron un mayor grado de respuesta durante 24 semanas cuando fueron comparados con sujetos con el genotipo TG/TG. Sobre la base de estos resultados, el genotipo 196TT estaría correlacionado con una mayor respuesta al tratamiento anti-TNF en la AR, mientras que el alelo G lo haría con una peor respuesta98.
La combinación del genotipo TNF -308 G/G y IL-10 -1082G/G (sujetos con una respuesta inflamatoria baja) también muestra una mejor respuesta al etanercept. Por tanto, el etanercept parece ser más efectivo en sujetos que portan un genotipo que codifica para una respuesta inflamatoria baja12. Otro estudio muestra polimorfismos microsatélites en la región promotora de la IL-10 asociados a una mejor respuesta al tratamiento a largo plazo con etanercept99.
También se han desarrollado diversos estudios farmacogenéticos sobre la eficacia del infliximab. La presencia del SNP -308 G/A en la región promotora del TNF influye en la respuesta a infliximab100–102; los sujetos que son portadores del genotipo G/G responden mejor al tratamiento. Algunos autores especularon sobre la posibilidad de que el polimorfismo TNF -308 podría influir en la respuesta a infliximab debido a los efectos que tiene sobre las concentraciones circulantes de TNF101; la presencia del alelo A (alto productor de TNF) puede tener relación con una peor respuesta a infliximab. Sin embargo, otros trabajos demuestran que no hay ninguna relación con la respuesta a infliximab11,98. En la misma línea, un estudio llevado a cabo por este grupo de investigación en 113 sujetos con AR mostró que los polimorfismos -308G/A y -238 G/A así como la presencia del epítopo compartido o del alelo DR3 no se correlacionan con una mejor respuesta a infliximab tras 30 semanas de tratamiento, según la cuantificación en base al índice DAS28103.
Hasta la fecha, únicamente hay un estudio farmacogenético en el que se analiza el grado de respuesta a la anakinra. En éste se pone de manifiesto la relación entre el alelo poco frecuente del polimorfismo IL-1α +4845 G/T y una respuesta significativa al fármaco104.
ConclusionesEn resumen, la respuesta al tratamiento está parcialmente determinada por la carga genética de un individuo. Como se ha indicado previamente, la AR es una enfermedad bien definida con un criterio ampliamente aceptado105, mientras que su aspecto clínico y las vías moleculares involucradas en el proceso son heterogéneos106. Por tanto, la respuesta a distintos tratamientos varía considerablemente entre los sujetos. Debido al desarrollo de una gran variedad de nuevos fármacos, sus precios y la falta de información detallada sobre sus efectos adversos así como la susceptibilidad a infecciones aumentan la necesidad de desarrollar marcadores genéticos que sean pronósticos en la respuesta frente al tratamiento107. Estos marcadores pueden encontrarse en los genes previamente descritos o también en los que codifican para las proteínas involucradas en la diana de acción del tratamiento, en su metabolismo o en la patogénesis de la enfermedad108. En este sentido, cabe destacar el articulo de Lequerré et al109, que obtuvieron a partir de células periféricas de sangre un perfil genético de 41 transcritos de ARNm susceptibles de predecir una respuesta a un tratamiento combinado de infliximab y MTX. De este modo, el entendimiento de la contribución genética en el tratamiento de la AR se convertirá en un hecho cada vez más relevante, en el momento en que las dianas de acción de los tratamientos desarrollados sean los propios mecanismos que desarrollan la enfermedad107.
Hasta la fecha se han descrito numerosos polimorfismos en diferentes genes, como el TNF, TNFRSF1B, alelos del MHC y otros genes de citocinas, pero su función es controvertida y algunos estudios ofrecen resultados contradictorios; son varias las causas propuestas para explicarlo, entre las que cabe destacar la estratificación de la población y el desequilibrio de ligamiento110. Además, el análisis de los haplotipos presentes en regiones de genes candidatos parece ser un método más adecuado que la descripción de SNP individuales. Sin embargo, como la farmacogenética es un campo relativamente nuevo, en el que los distintos trabajos están empezando a publicarse, se puede especular con que, en un futuro tal vez no muy lejano, pueda ser posible aplicar un tratamiento personalizado en función del genotipo de un individuo30. Para conseguir esto se requieren grandes estudios que involucren a múltiples instituciones con el fin de obtener el número adecuado de sujetos para determinar si las variantes génicas descritas en los genes de las citocinas, así como de otras moléculas especificas, contribuyen directamente tanto en la patofisiología como en la respuesta a diversos tratamientos de la AR.
Los autores de este artículo agradecen a la Srta. P. Cal Purriños su experta asistencia como secretaria.
Conflicto de intereses
Ninguno.


