




Las vasculitis sistémicas primarias (VSP) constituyen un grupo heterogéneo de procesos clínicos complejos y graves con un sustrato patológico común: inflamación y necrosis de los vasos sanguíneos. En el proceso inflamatorio se ven afectados vasos de muy diferentes tamaños. La localización y el diferente tamaño de los vasos afectados, la gravedad del daño vascular y el distinto patrón histopatológico constituyen características que definen los diferentes síndromes vasculíticos y permiten su individualización. La descripción y el avance en el conocimiento de las vasculitis primarias han evolucionado considerablemente en las últimas décadas. Esta revisión tiene como objetivo ofrecer un punto de vista respecto del abordaje práctico de estas enfermedades y su dificultad. Se centra más en las vasculitis asociadas con anticuerpos contra citoplasma de neutrófilos (VAA), debido a que este subgrupo es el que tiene el único biomarcador comprobado en este grupo de enfermedades, es aparentemente el más frecuente en nuestro medio y el que mayor afectación multiorgánica puede tener. Se hacen apuntes relativos a otras VSP también.
The primary systemic vasculitidies (PSV) comprise an heterogeneous group of complex clinical entities with a common substrate: inflammation and necrosis of blood vessels. The inflammatory process involves vessels of any caliber. The location and the different size of the affected vessels, the severity of vascular damage and the different histopathological patterns which may predominate are the basic characteristics that define the different vasculitic syndromes and enable individualization. The description and progress in the knowledge of the primary vasculitidies have evolved considerably in recent decades, allowing for a better resolution of the complex problems placed by these patients. This review aims to provide a view on the practical approach to these diseases and their challenge. It focuses on ANCA associated vasculitidies (AAV) because this subgroup has the only proven biomarker and is the most common in our area. Also, multiorganic involvement is frequent. Some notes regarding other PSV are made.
Las vasculitis asociadas a anticuerpos contra citoplasma de neutrófilos (ANCA), las cuales se denominarán VAA en adelante, comprenden la granulomatosis con poliangeítis (Wegener) (GPA), la poliangeítis microscópica (PAM) y el síndrome de Churg-Strauss (SCS).
La epidemiología de las VAA plantea retos considerables para los epidemiólogos. El primero es la dificultad de definir un caso individualmente, dada la presencia ocasional de distinción claramente inequívoca entre los diferentes trastornos. Hay dos sistemas principales de la definición de casos o clasificación, de uso común en la literatura, el del Colegio Americano de Reumatología (ACR) (1990)1 y los criterios de clasificación del Consenso Chapel Hill (CHCC)2. Hay varios problemas con estos criterios cuando se utilizan con fines epidemiológicos. La PAM no figura en el sistema ACR, pero, en contraparte, el CHCC tampoco toma en cuenta a los ANCA. El CHCC define algunas vasculitis y los criterios propuestos no pueden utilizarse como clasificatorios. Por lo tanto, en este momento no hay criterios validados de clasificación para la PAM. Para superar esto, es frecuente que ambos sistemas se usen en forma paralela, lo que ha llevado a superposición considerable entre distintas categorías3. En un intento de superar esta situación, un grupo internacional de consenso propuso un algoritmo, que incorpora distintos sistemas y ha sido validado en dos poblaciones separadas4,5. La segunda dificultad es la captura de casos. Las VAA son raras y se necesita una gran población para determinar la incidencia y la prevalencia, lo que plantea dificultades en su viabilidad. La tercera dificultad es la verificación de casos; por su infrecuencia, en ocasiones solamente se refieren y corroboran en centros especializados. Sin embargo, los pacientes con enfermedad fulminante pueden morir antes del diagnóstico y sin comprobarse la enfermedad. La rareza de estos síndromes hace que los estudios prospectivos de casos y controles sean difíciles porque el tamaño de la población requerida para obtener una estadística confiable no está fácilmente disponible. A pesar de lo anterior, se ha recopilado un volumen considerable de datos sobre la epidemiología de las VAA en los últimos 20 años, aunque gran parte de los datos proceden de las poblaciones de ascendencia europea. Existe un amplio consenso en que las vasculitis sistémicas primarias (VSP), de medianos y pequeños vasos (incluidos GPA, SCS, poliarteritis nodosa [PAN] y PAM]), tienen una incidencia global anual de aproximadamente 10 a 20/millón, con edad pico de aparición de 65 a 74 años6-11.
Un aspecto fundamental para conocer mejor estos síndromes ha sido la introducción de los ANCA como marcadores de las vasculitis de vasos pequeños12,13, pero ello no ha sido el caso en otras VSP.
De este modo, no resulta extraño que el diagnóstico oportuno de las VSP constituya uno de los mayores retos que se le pueden presentar al clínico.
¿Qué rutas diagnósticas seguir en un paciente en quien se sospecha una vasculitis sistémica primaria?En estas patologías nunca sobra enfatizar enérgicamente que la evaluación inicial debe incluir una cuidadosa y completa historia clínica, aunada a un minucioso examen físico como primera ruta diagnóstica. En muchas ocasiones, los pacientes presentan síntomas generales, entre los que se encuentran mialgias, artralgias, artritis (fig. 1), e incluso fiebre prolongada de origen desconocido (mayor a 38°C). Estas manifestaciones son absolutamente inespecíficas y, por tanto, proveen poca certeza inicial. A esto pueden agregarse alteraciones que traducen afectación multisistémica, como dolor abdominal, daño cardiovascular y alteraciones del sistema nervioso central o periférico. Sin embargo, la aparición de otras manifestaciones, como insuficiencia renal rápidamente progresiva, acompañada de hipertensión arterial, alteraciones del aparato respiratorio en forma de asma, sinusitis repetida, neumonía o la presencia de infiltrados pulmonares inexplicables, lesiones cutáneas como púrpura palpable, úlceras no infecciosas nodulaciones, o bien alteraciones del sistema nervioso periférico como mononeuritis múltiple o polineuropatía, hacen más viable la probabilidad de que una VSP sea la responsable de estos síntomas cuando se presentan en un individuo. Sin embargo, ello puede aparecer de forma gradual en el lapso de varias semanas o meses, y mientras tanto, el paciente ha sido ya tratado con la sospecha de otra enfermedad, lo cual puede modificar la presentación y, por tanto, el retraso de un tratamiento adecuado.
.")
Hay ciertas manifestaciones clínicas que pueden orientar más hacia una ruta diagnóstica adecuada. En los pacientes con VAA, la presencia de lesiones granulomatosas necrosantes en el tracto respiratorio superior y/o inferior apunta a GPA. A pesar de ello, muchas veces esto incluso tarda en reconocerse, ya que entidades o síntomas como sinusitis crónica, rinitis crónica, ulceración nasal, epistaxis, otitis media serosa, tos, hemoptisis y disnea pueden ser vistos por especialistas dedicados a estos aspectos por meses o años antes de que otras manifestaciones se hagan presentes y se revele la característica afectación multiorgánica de esta enfermedad. En relación con esta, la GPA, en términos generales, es más común en los hombres, con un intervalo de edad de 8 a 80 años y una edad media de 40 años; en el 80% de los casos puede presentarse afectación renal durante la evolución de la enfermedad, generalmente posterior a manifestaciones respiratorias altas y a veces bajas también. Se caracteriza por hematuria, proteinuria y distintos grados de insuficiencia renal; la biopsia muestra glomerulonefritis necrosante segmentaria con mínimo o ningún depósito de inmunoglobulinas o complemento en la inmunofluorescencia, y clínicamente se comporta en ocasiones de forma rápidamente progresiva. Hasta en el 17% de los casos es la manifestación inicial. Una característica adicional de este afección es la afectación ocular, en forma de seudotumor orbitario, escleritis, queratitis o uveítis. Respecto de las manifestaciones neurológicas, de un 35 a un 50% de los pacientes las tiene. En su mayoría son del sistema nervioso periférico, donde casi la mitad tiene mononeuropatía recurrente, mononeuritis múltiple o polineuropatía simétrica. Los pacientes con compromiso del sistema nervioso central (SNC) se pueden presentar con neuropatías craneales, con mayor frecuencia de los pares II, VI y VII, hemiparesia, convulsiones, afasia y defectos del campo visual, así como vasculitis del SNC. Son menos frecuentes las apoplejías isquémicas, la encefalopatía, la meningitis granulomatosa basilar, la encefalopatía, la paquimeningitis o la mielitis. La principal especificidad antigénica reconocida es contra proteinasa-3 (PR-3), responsable del patrón citoplasmático de los ANCA en hasta el 90% de los casos, particularmente aquellos con afectación general14,15.
En cuanto a la PAM, esta es una vasculitis necrosante que afecta a los vasos de pequeño calibre y que carece de inflamación granulomatosa, lo que la distingue de la GPA; en la PAM, las alteraciones renales son más frecuentes, por ende, de manera comparativa con la GPA, la glomerulonefritis rápidamente progresiva, con necrosis glomerular focal y segmentaria es más común. El promedio de edad de inicio es de 50 años y es un poco más frecuente en los hombres. Los síntomas de afectación constitucional. como artralgias o fiebre, pueden estar presentes mucho antes del reconocimiento de la enfermedad, cuando se presenta como nefritis rápidamente progresiva. La afectación pulmonar ocurre con mayor frecuencia que en las vasculitis de mediano calibre. Los problemas neurológicos son también frecuentes y en nuestros pacientes (datos no publicados) se aprecia neuropatía periférica en hasta el 75% de los casos. La principal especificidad antigénica de los ANCA se dirige contra la mieloperoxidasa (MPO)16. El SCS tiene desde luego ciertos antecedentes y hallazgos importantes, de los que se enfatizan el asma y la eosinofilia periférica. En esta enfermedad es importante reconocer la afectación gastrointestinal, y particularmente la cardíaca, que es la primera causa de muerte en estos pacientes. La positividad de ANCA en esta entidad es la menos frecuente de las tres17,18.
La PAN fue la primera VSP descrita. Afecta particularmente a los vasos arteriales de mediano calibre; es importante reconocer que la forma primaria se ha vuelto extremadamente poco prevalente en las últimas décadas y que muchos de los casos antes observados se asocian a otras afecciones, particularmente hepatitis B (y en menor frecuencia C) e infección por el VIH, con algunos otros relacionados con leucemia. Puede afectar a cualquier órgano; sin embargo, la piel, los nervios periféricos, el tracto gastrointestinal y los riñones (por isquemia de vasos de mayor calibre a los glomérulos) son los más comúnmente afectados. La revisión de grandes series muestra que la edad promedio de los pacientes está alrededor de los 50 a 60 años y que, en comparación con las formas primarias, los pacientes con PAN secundaria a una infección por virus de hepatitis B tienen con mayor frecuencia neuropatía periférica, dolor abdominal, cardiomiopatía, orquitis, hipertensión y afectación neurológica (en particular, la mononeuritis múltiple)19. Debemos hacer mención especial respecto de que, a la fecha, es claro que esta vasculitis no se asocia con ANCA y que muchos de los casos que así se describieron inicialmente correspondían a pacientes con PAM.
En contraste con las VAA, la púrpura de Schönlein-Henoch es más común en pacientes pediátricos y se caracteriza por afectación cutánea, articular, del tracto gastrointestinal, y renal; esta última muestra una histología similar a la vista en la glomerulonefritis asociada con ANCA, aunque con el característico depósito de IgA en los glomérulos mediante inmunofluorescencia. Algunos pacientes presentan manifestaciones neurológicas periféricas, si bien ello es raro20.
En nuestro medio, las dos vasculitis de vasos de gran calibre son más infrecuentes que aquellas VAA, particularmente la arteritis de células gigantes (ACG), que es casi inexistente. En estas patologías, un aspecto fundamental es el grupo de edades y características raciales de quienes las desarrollan; la ACG predomina claramente en caucásicos y en pacientes mayores de 60 años, mientras que la arteritis de Takayasu lo es en pacientes de origen oriental, mujeres, y menores de 40 años. En todo caso, las manifestaciones isquémicas en ambas vasculitis son muy importantes e incluyen claudicación de miembros torácicos y pélvicos, de mandíbula, dolor facial, síntomas neurológicos de origen central, manifestaciones oculares que representan isquemia tales como amaurosis fugax, diplopía o defectos del campo visual, además de manifestaciones generales que se han descrito antes en las otras VSP, pero cuya forma de presentación puede ser incluso más prolongada e indolente. Un aspecto esencial en la evaluación de los pacientes en quienes se sospechan estas dos entidades es la palpación de los pulsos periféricos para evaluar si existe asimetría y la presencia de frémito, la evaluación de la presión arterial en todas las extremidades y la auscultación cuidadosa en búsqueda de soplos21,22.
Dentro de la ruta diagnóstica, los estudios paraclínicos (serológicos e histopatológicos) y de imagenología útiles para el diagnostico de las VSP se describen a continuación. Los exámenes de laboratorio deben incluir un hemograma completo, química sanguínea, examen general de orina, con análisis minucioso del sedimento urinario y, si el examen general de orina muestra proteinuria aislada, se debe efectuar cuantificación de proteínas en orina de 24 h, depuración de creatinina y pruebas de función hepática, así como medición de los reactantes de fase aguda (proteína C reactiva y velocidad de sedimentación globular). Los ANCA han demostrado ser de gran utilidad y un biomarcador sensible y específico23 de uno de los subgrupos más frecuentes de VSP siempre y cuando se hagan en laboratorios confiables y en un contexto clínico apropiado. No es objeto de esta revisión describir en detalle las características de esta prueba, para lo que se refiere al lector a excelentes revisiones12; sin embargo, es importante mencionar que no todos los pacientes con vasculitis tienen ANCA positivos.Los informes sobre los ANCA en las diferentes formas de VAA varían considerablemente. Se ha informado su positividad en el 40 al 95% de los pacientes con GPA, el 40-90% de los sujetos con MPA y en el 10 al 70% de los pacientes con SCS12,13,15-18. La razón de estas discrepancias se puede deber a las cohortes seleccionadas, la extensión de la enfermedad y la actividad en el momento de toma de las muestras, los criterios utilizados para establecer el diagnóstico y los valores límite positivos y puntos de corte establecidos de los ensayos utilizados. Este problema no ha sido suficientemente aclarado por los productores de técnicas comerciales de ELISA o por los laboratorios diagnósticos que utilizan estuches comerciales. Con fines de utilidad de los clínicos que deciden entre varios diagnósticos diferenciales, sería necesario no solo un valor de corte positivo de ANCA en donantes sanos, sino también en pacientes con diferentes enfermedades inflamatorias.
Entre médicos de primer contacto y otros no habituados por su especialidad a ver a pacientes con estos problemas, esta es una premisa inicial fundamental. El resultado positivo de ANCA no es sinónimo de vasculitis, como lo inverso tampoco, es decir, que las vasculitis, ya sea primarias o secundarias, deban ser ANCA positivas. Indicar en exceso esta prueba en contextos clínicos poco sustentables de una vasculitis primaria puede resultar grave por las consecuentes decisiones cuando un paciente resulta positivo en una prueba sin tener VAA o, por el contrario, descartarla solamente basándose en el resultado de los ANCA, sin tomar en cuenta todos los elementos apropiados24.
Por otro lado, en este momento, solo están claramente establecidos el vínculo y la solidez de este cuando los ANCA detectados por inmunofluorescencia indirecta (IFI) tienen como antígenos blanco a la PR-3 y la MPO23,25. Muchos otros antígenos contenidos en los neutrófilos y monocitos pueden dar positividad en esta prueba mediante IFI y no ser vasculitis. Como ejemplo más común, los P-ANCA están presentes en otras enfermedades inflamatorias crónicas, como la artritis reumatoide, el síndrome de Felty, la colitis ulcerosa crónica, la hepatitis crónica, la cirrosis biliar primaria, la colangitis esclerosante y el síndrome de Sweet, entre otros. En común, todos ellos reconocen en forma frecuente otros antígenos distintos a PR-3 y MPO. Por lo tanto, la comprobación mediante estudios sobre fase sólida como el ELISA, acerca de su especificidad contra PR-3 y/o MPO es necesaria, con el fin de descartar que dicho resultado por IFI se explique al reconocer antígenos que no se han asociado en su autorreactividad con las VAA25.
Otra mención importante se refiere a la presencia de vasculitis secundarias, como las vistas en otras enfermedades autoinmunitarias, como el lupus eritematoso sistémico, las asociadas a ciertos fármacos, en particular los medicamentos antitiroideos, y aquellos sujetos con destrucción nasal por cocaína. En estos pacientes, pueden existir ANCA positivos por IFI a títulos altos, pero los antígenos que reconocen pueden ser diferentes, como la elastasa, en el caso de los usuarios de cocaína, o la MPO, que puede verse en las vasculitis asociadas a lupus o a fármacos; la polirreactividad a otros antígenos, que explica la coexistencia de otros marcadores serológicos positivos (anticuerpos contra ADN, cardiolipina, beta-2-glucoproteína I), es frecuente y, desde luego, es muy importante la presencia de ciertos datos clínicos propios de la otra patología autoinmunitaria o el antecedente de exposición a fármacos, así como la inconsistencia entre los hallazgos histológicos y las manifestaciones clínicas (véase más adelante). Por tales razones, hay autores que han propuesto que estos otros autoanticuerpos sean denominados de otra forma y no ANCA26,27.
Algo que se debe recordar es que un pequeño porcentaje de pacientes con demostración histológica de VAA son ANCA negativos. Otras pruebas serológicas recomendadas dependen de la evaluación inicial, pero dado que muchas entidades autoinmunitarias comparten varios de los síndromes clínicos presentes en las VSP, puede decirse, en forma general, que es conveniente solicitar anticuerpos antinucleares (AAN) en células HEp-2, complemento y factor reumatoide. De acuerdo con la positividad y patrón de los AAN, puede solicitarse serología específica (anti-ADN, anti-Sm, anti-RNP, anti-Scl70, anti-Ro, anti-La, anticentrómero, etc.). En caso de sospecharse una causa infecciosa de la vasculitis sospechada, se deben solicitar la serología contra el VIH o el virus de las hepatitis B o C, así como las crioglobulinas.
Los estudios de imagenología representan una importante herramienta. Por ejemplo, una radiografía de tórax es adecuada en la evaluación inicial, dada la afección pulmonar frecuente en la GPA, en ocasiones en forma silente si se trata de enfermedad nodular que traduce inflamación granulomatosa. Otros ejemplos de ello se dan en la reciente descripción de casos de fibrosis pulmonar idiopática en PAM, la afectación cardiaca en el SCS y la arteritis de Takayasu (en forma secundaria, muchas veces como resultado de hipertensión arterial o de valvulopatía). Por otro lado, avances recientes en imagenología son particularmente útiles en el caso de las vasculitis de grandes vasos, lo que permite evitar angiografías convencionales. Este aspecto se detalla en otro artículo de este número. Estudios adicionales de gabinete irán de acuerdo con las entidades específicas y se pueden emplear en la investigación del diagnóstico diferencial en las vasculitis de pequeños vasos; la tomografía computarizada (TC) y la resonancia magnética proporcionan información útil sobre la inflamación y el daño de órganos, como en el caso de la GPA, en la que la TC de senos paranasales muestra datos característicos. Una TC de alta resolución (TACAR) de los pulmones es fundamental en las VAA.
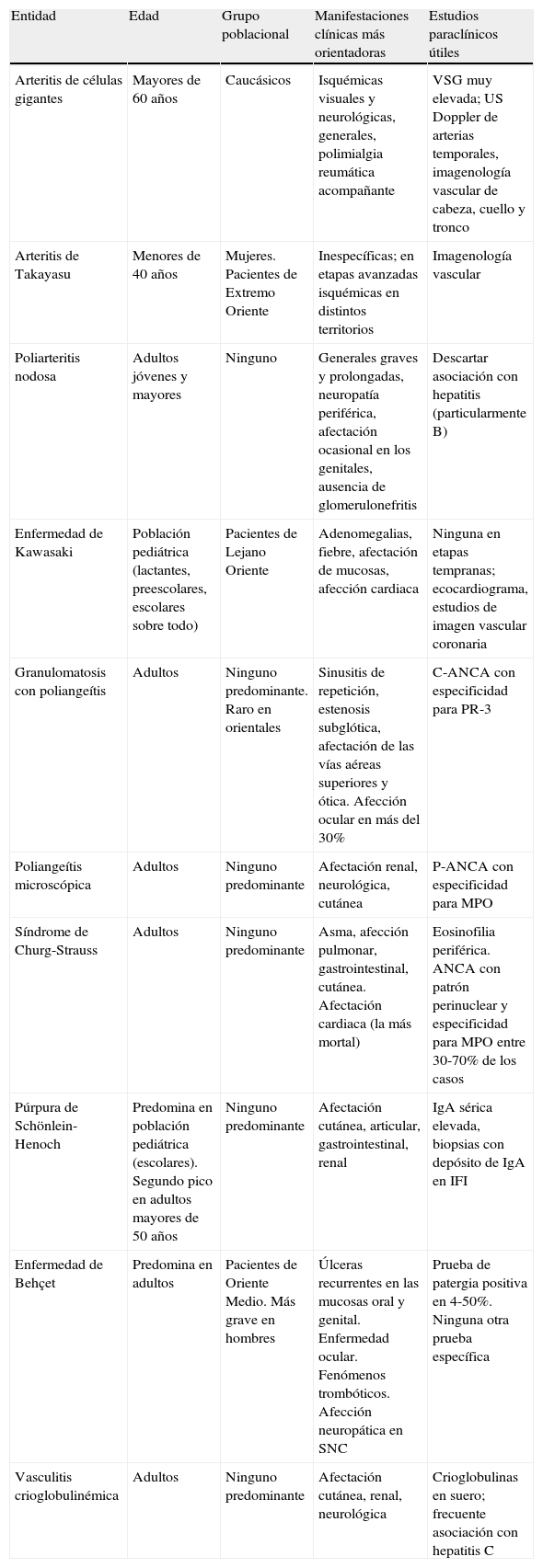
En la tabla 1 se resumen ciertos aspectos de orden demográfico y clínico que pueden ser orientadores como puntos que se deben considerar al sospechar alguna VSP y, en particular, se presentan los que en forma práctica orientan a alguna de ellas.
Ciertas características de orden práctico presentes en las VSP que permiten orientar cuál entidad es la presente
| Entidad | Edad | Grupo poblacional | Manifestaciones clínicas más orientadoras | Estudios paraclínicos útiles |
| Arteritis de células gigantes | Mayores de 60 años | Caucásicos | Isquémicas visuales y neurológicas, generales, polimialgia reumática acompañante | VSG muy elevada; US Doppler de arterias temporales, imagenología vascular de cabeza, cuello y tronco |
| Arteritis de Takayasu | Menores de 40 años | Mujeres. Pacientes de Extremo Oriente | Inespecíficas; en etapas avanzadas isquémicas en distintos territorios | Imagenología vascular |
| Poliarteritis nodosa | Adultos jóvenes y mayores | Ninguno | Generales graves y prolongadas, neuropatía periférica, afectación ocasional en los genitales, ausencia de glomerulonefritis | Descartar asociación con hepatitis (particularmente B) |
| Enfermedad de Kawasaki | Población pediátrica (lactantes, preescolares, escolares sobre todo) | Pacientes de Lejano Oriente | Adenomegalias, fiebre, afectación de mucosas, afección cardiaca | Ninguna en etapas tempranas; ecocardiograma, estudios de imagen vascular coronaria |
| Granulomatosis con poliangeítis | Adultos | Ninguno predominante. Raro en orientales | Sinusitis de repetición, estenosis subglótica, afectación de las vías aéreas superiores y ótica. Afección ocular en más del 30% | C-ANCA con especificidad para PR-3 |
| Poliangeítis microscópica | Adultos | Ninguno predominante | Afectación renal, neurológica, cutánea | P-ANCA con especificidad para MPO |
| Síndrome de Churg-Strauss | Adultos | Ninguno predominante | Asma, afección pulmonar, gastrointestinal, cutánea. Afectación cardiaca (la más mortal) | Eosinofilia periférica. ANCA con patrón perinuclear y especificidad para MPO entre 30-70% de los casos |
| Púrpura de Schönlein-Henoch | Predomina en población pediátrica (escolares). Segundo pico en adultos mayores de 50 años | Ninguno predominante | Afectación cutánea, articular, gastrointestinal, renal | IgA sérica elevada, biopsias con depósito de IgA en IFI |
| Enfermedad de Behçet | Predomina en adultos | Pacientes de Oriente Medio. Más grave en hombres | Úlceras recurrentes en las mucosas oral y genital. Enfermedad ocular. Fenómenos trombóticos. Afección neuropática en SNC | Prueba de patergia positiva en 4-50%. Ninguna otra prueba específica |
| Vasculitis crioglobulinémica | Adultos | Ninguno predominante | Afectación cutánea, renal, neurológica | Crioglobulinas en suero; frecuente asociación con hepatitis C |
PR-3: proteinasa 3; MPO: mieloperoxidasa; VSG: velocidad de sedimentación globular; VSP: vasculitis sistémica primaria.
En el contexto de la ruta diagnóstica, el examen histológico es considerado el estándar de oro en las vasculitis. A pesar de ello, no siempre se obtiene el diagnóstico. Por ejemplo, en el caso de la biopsia de la arteria temporal, puede existir un 10% de pacientes en quienes no se hallan las características histopatológicas conocidas, incluso en centros de excelencia. La naturaleza segmentaria de la enfermedad explica en parte esto28. De igual forma, en un cierto subgrupo de pacientes con lesiones histológicas características en biopsias renales29 o cutáneas (figs. 2 y 3), los hallazgos pueden no correlacionar del todo con los resultados de los ANCA. En todo caso, las manifestaciones clínicas, apoyadas en lo posible por métodos de imagen que muestren afección de determinados órganos en forma más objetiva, pueden guiar la obtención de tejido con fines confirmatorios. Por ejemplo, la biopsia renal es particularmente útil en el diagnóstico de las VAA y la exclusión de otras enfermedades, como neoplasias o infecciones. Además, las características histológicas proporcionan oritentación pronóstica en la glomerulonefritis asociada a ANCA30. Por otra parte, la utilidad de la biopsia se extiende en algunos casos a demostrar la presencia de cicatrización con daño funcional. También, dada la ausencia de biomarcadores más específicos que correlacionen adecuadamente con el grado de actividad inflamatoria de la enfermedad, el examen histológico puede ser el único medio de exclusión de inflamación activa y puede guiar las decisiones terapéuticas. Si bien siempre será deseable obtener tejido histológico para la confirmación del padecimiento sospechado, algunas condiciones no siempre lo permiten. Por ejemplo, en un paciente con nódulos pulmonares, que se encuentre en ventilación mecánica, con diátesis hemorrágica e inestabilidad hemodinámica, la obtención de tejido adecuado para la confirmación histológica es difícil. En este contexto, la exclusión por todos los medios posibles de otras condiciones que expliquen la manifestación clínica que hizo sospechar una vasculitis y la indicación, realización correcta, así como la adecuada interpretación de las pruebas serológicas, como los ANCA, pueden ser clave para la toma de decisiones inmediatas, lo que permite esperar un mejor momento para obtener confirmación histológica, la cual es deseable en casos complejos.
. Estas lesiones pueden verse prácticamente en cualquier caso de vasculitis primaria de vasos de pequeño calibre. Obsérvese cómo algunas lesiones coalescen y otras tienen centro necrótico.")
. La biopsia de la piel afectada mostrada en la imagen anterior presenta en el panel superior, infiltrado en la dermis papilar, con extensión a la dermis reticular (40×) y un acercamiento, en el panel inferior, del infiltrado que ocupa toda la pared del vaso sanguíneo, con leucocitoclasia (100×, HE).")
A y B). La biopsia de la piel afectada mostrada en la imagen anterior presenta en el panel superior, infiltrado en la dermis papilar, con extensión a la dermis reticular (40×) y un acercamiento, en el panel inferior, del infiltrado que ocupa toda la pared del vaso sanguíneo, con leucocitoclasia (100×, HE).
Finalmente, debemos citar que una biopsia negativa no excluye necesariamente la enfermedad y una biopsia con cambios compatibles, pero explicados satisfactoriamente por otras patologías, como puede ser la presencia de inflamación vascular en un foco de infección fúngica o micobacteriana, no siempre indica la presencia de una VSP, por lo que es necesaria la adecuada correlación clínico-patológica de estas enfermedades para evaluar la evolución de los pacientes.
¿Cómo hacer el diagnóstico diferencial entre las vasculitis sistémicas primarias y las secundarias a otras patologías?Ya que no es sorprendente que muchas enfermedades de diversa causa constituyan parte esencial del diagnóstico diferencial de las VSP, es importante conocer las condiciones que regionalmente puedan ser importantes para un ejercicio diferencial adecuado. Ya que las implicaciones terapéuticas de las VSP pueden conllevar un riesgo de adquirir o exacerbar infecciones, estas emergen como las más importantes que se deben considerar. Como ejemplo valga citar que padecimientos infecciosos crónicos, como micobacteriosis o infecciones por hongos, pueden dar síntomas similares, con extensión multiorgánica e incluso serología positiva para ANCA. Así, en países como el nuestro, o algunos lejanos, como India, el contexto clínico y serológico de sujetos con fiebre, lesiones cutáneas y manifestaciones pulmonares debe ser adecuadamente considerado31,32. Además de estas condiciones y de otras, como las enfermedades neoplásicas, que son grandes simuladoras, existe un grupo importante de condiciones que tienen como órgano blanco fundamental la piel y que, en forma global, se conocen como seudovasculitis. Estas lesiones, que se pueden manifestar clínicamente como petequias, púrpura, equimosis, úlceras digitales, necrosis, gangrena, nódulos o úlceras, en muchas ocasiones recurrentes, obligan a la minuciosa búsqueda de factores condicionantes de su presencia.
Ejemplos de estas seudovasculitis son: seudovasculitis hemorrágica, que se produce por la incompetencia de las paredes de los vasos afectados y puede estar relacionada con diversos factores que incluyen el depósito en la pared vascular de sustancias (amiloide, calcio), deficiencias nutricionales (escorbuto), púrpura inflamatoria no vasculítica (dermatitis purpúrica pigmentada), por artrópodos, virus o reacciones a medicamentos, así como los trastornos de la coagulación como la trombocitopenia. La seudovasculitis cianótica se debe a la oclusión de un vaso sanguíneo por émbolos, trombos o hiperplasia fibrointimal (endarteritis obliterante) e incluye diversas condiciones, tales como púrpura fulminante, necrosis por cumarínicos, síndrome por anticuerpos antifosfolípido, mixoma cardiaco, embolización por colesterol, calcifilaxis y arteritis por radiación33,34.
ConclusionesEl diagnóstico de las VSP es una de las más difíciles tareas a las que cualquier clínico puede enfrentarse. La presencia de una amplia constelación de hallazgos clínicos, serológicos, histológicos y de imagen debe considerarse en su justo peso después de un ejercicio clínico completo. Una apropiada orientación diagnóstica, que considere los diagnósticos diferenciales más adecuados a cada paciente particular y su entorno, es la piedra angular para el diagnóstico oportuno. Estas dificultades son un reto para cualquier médico, como lo han sido para inmunólogos y epidemiólogos, si bien cada vez más hay interés en estas enfermedades, lo que ha permitido un avance en su conocimiento.
La nomenclatura de estas enfermedades está incluso siendo revisada. No hay duda de que el aumento en el conocimiento de los mecanismos fisiopatogénicos y, en un futuro, de la causa de algunas de ellas permitirá, como ha sucedido recientemente con la granulomatosis de Wegener, ahora denominada GPA (Wegener), una mejor definición de estas patologías.
Otro reto es la carencia de criterios diagnósticos en las VSP, aunque actualmente está en curso un proyecto de investigación multinacional para elaborarlos. Los actuales criterios de clasificación del ACR fueron desarrollados en 1990, hace 21 años, antes de la disponibilidad de algunas pruebas de diagnóstico muy importantes (p. ej., ANCA). Esfuerzos multinacionales, como el proyecto conjunto de ACR y EULAR (Liga Europea en contra del Reumatismo), llamado «Estudio sobre el diagnóstico y clasificación de las vasculitis» (DCVAS por sus siglas en inglés)35, buscan solventar este aspecto en los próximos años.
El advenimiento y el posicionamiento de los ANCA en el diagnóstico de las VSP representaron un gran avance en el diagnóstico, pero ello solamente ha permitido la mejor caracterización de un pequeño número de estas enfermedades y, en todo caso, tampoco han permitido agregar valor en el seguimiento anticipado de pacientes en momentos de recurrencia de la enfermedad o en discriminar la coexistencia de complicaciones como infecciones, comunes en el curso del tratamiento de la GPA o la PAM. El reto en encontrar otros biomarcadores para otras VSP está en pie y su búsqueda es una tarea continua.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


