









La enfermedad relacionada con IgG4 (ER-IgG4) es una entidad recientemente nominada para definir diversas enfermedades caracterizadas por infiltración linfoplasmocítica, fibrosis, presencia de un número aumentado de células IgG4+ y, en gran parte de los casos, niveles aumentados de IgG4 sérica, afectando frecuentemente el páncreas, las glándulas salivales y los ganglios linfáticos pero pudiendo comprometer casi cualquier estructura de la anatomía humana. Aunque su etiología se desconoce, se han realizado avances en el conocimiento de sus bases fisiopatológicas e inmunológicas, al igual que del rol de las células inflamatorias en el desarrollo de daño del órgano blanco. No existe hasta la fecha un consenso internacional sobre su diagnóstico, lo que no ha impedido avances terapéuticos muy importantes en su control y búsqueda de remisión. Se hace una revisión acerca de la historia, hipótesis sobre la etiología de la enfermedad, sus manifestaciones clínicas, abordaje diagnóstico y terapéutico.
IgG4-related disease is the term used to refer to a condition characterized by a lymphoplasmacytic infiltrate, fibrosis and an increased number of IgG4+ cells present in tissue, in most cases, with an elevated serum IgG4 level. This disease frequently affects the pancreas, salivary glands and lymph nodes, but can involve almost any tissue. Its etiology and the exact role of the different inflammatory cells in the damage to the target organ is still unclear. As yet, there is no international consensus about diagnostic criteria for the disease, but there are important advances in its treatment and in the quest to achieve remission. We include a review of the history, possible pathogenesis, clinical manifestations, diagnostic approach and available therapeutic approaches.
Se realizó una revisión no sistemática de la literatura en idiomas inglés y español en las bases de datos Pubmed y Scielo, con el objetivo de describir aspectos fundamentales sobre la enfermedad relacionada con IgG4 (ER-IgG4): descripción histórica inicial de la enfermedad y evolución de la misma, bases fisiopatológicas e inmunológicas, manifestaciones clínicas, diagnóstico y aproximación terapéutica actual. En ambas bases de datos se seleccionaron artículos publicados en los últimos 10años. En Pubmed se empleó la estrategia de búsqueda (Immunoglobulin g4 [MeSH] related disease), arrojando un total de 132 resultados. En Scielo se empleó la combinación (Igg4 related disease) sin encontrar otros resultados pertinentes diferentes a los obtenidos en Pubmed. Se hizo búsqueda puntual de las referencias históricas mencionadas en los artículos encontrados con la estrategia anterior al igual que de artículos de interés mencionados en las bibliografías respectivas. En total se incluyeron 49 referencias: 42 provenientes de la búsqueda inicial y 7 obtenidas a partir de las bibliografías.
Introducción y recuento históricoLa enfermedad relacionada con IgG4 (ER-IgG4) es la denominación otorgada durante la década anterior a una entidad caracterizada por lesiones tumefactas, un infiltrado denso linfoplasmacítico con abundantes células positivas para IgG4, fibrosis estoriforme y frecuentemente, pero no siempre, niveles séricos elevados de IgG41. Las primeras descripciones de procesos patológicos compatibles con la enfermedad datan de 1892, cuando Johann von Mikulicz-Radecki describió un paciente de 42años, granjero, sin síntomas secos, quien presentaba «edema simétrico de las glándulas lacrimales, parótidas y submandibulares, con infiltración masiva de las mismas por células mononucleares» y que fallecería al año de esta descripción, permitiendo al Dr. Mikulicz realizar su autopsia2. En ella confirmó el aumento de tamaño de las glándulas mencionadas, múltiples adenopatías y el compromiso microscópico por un infiltrado mononuclear. El dibujo esquemático de lo que vio a través del microscopio asemeja en gran medida a un linfoma tipo MALT, pero es actualmente imposible confirmar uno u otro diagnóstico (o el desarrollo del linfoma sobre una ER-IgG4), obligando al clínico moderno a considerar siempre malignidad como un diagnóstico diferencial3.
El síndrome de Mikulicz cursa con parotidomegalia y posible aumento de glándulas submandibulares. Desde 1953 fue incluido como una de las posibles manifestaciones clínicas del síndrome de Sjögren4. Años después, en la década de los sesenta, se describieron casos de otra entidad denominada «pancreatitis crónica esclerosante»5. Algunos de estos casos incluían descripciones de un infiltrado linfoplasmocitario en el órgano afectado con aumento difuso del tamaño del mismo pero de causa desconocida6. En 1995 se propuso el concepto de pancreatitis autoinmune basándose en la comunicación de casos de pancreatitis con hipergammaglobulinemia, autoinmunidad positiva y respuesta a corticoides7, pero no sería hasta el año 2001 cuando se documentarían niveles séricos elevados de IgG4 en pacientes con pancreatitis esclerosante, separando así una nueva entidad nosológica8. En el año 2004 la documentación de niveles elevados de IgG4 en pacientes con enfermedad de Mikulicz la establecerían definitivamente dentro del espectro de ER-IgG49. La nomenclatura actual de ER-IgG4 se propuso en el año 2010 y fue aceptada en el 2011 durante el primer congreso internacional sobre la entidad, realizado en Boston, a partir del cual ha aumentado progresivamente el número de publicaciones anuales en la materia, incluyendo en 2012 el primer consenso internacional sobre los hallazgos patológicos que la definen actualmente10.
Incidencia, prevalencia y órganos comprometidosTres hallazgos histopatológicos caracterizan a la enfermedad en el órgano afectado: 1)la presencia de esclerosis de patrón estoriforme; 2)un denso infiltrado linfoplasmocitario, y 3)una proporción aumentada de células positivas para IgG4 por inmunohistoquímica respecto a las positivas para IgG. El promedio de esta proporción es una relación de células IgG4+/IgG+ mayor al 40%, pero el criterio varía según el órgano afectado10. Utilizando dichos hallazgos histológicos, se estimó que la prevalencia de la ER-IgG4 en Japón es de 2,63-10,2 casos por millón de habitantes, con una incidencia de 336-1.300 casos nuevos por año. Hasta la fecha se han publicado 5 cohortes de pacientes a partir de las cuales se puede extraer información sobre la presentación e historia natural de la enfermedad (tabla 1).
Cohortes publicadas de pacientes con ER-IgG4
| Autor(ref) | País | Pacientes (n) | Promedio de edad en años (rango)a | Sexo masculino | Manifestaciones más frecuentes u órganos comprometidos (%) | Desenlaces de los pacientesb |
|---|---|---|---|---|---|---|
| Ebbo et al.22 | Francia | 25 | 58 (24-83) | 72% | Adenopatías (76), pancreatitis esclerosante (52), sialadenitis (44), nefritis intersticial (44), colangitis esclerosante (32) | 92% tratados con corticoides, mejoría clínica en 90% de ellos. 48% con dependencia a corticoides o efectos secundarios de los mismos. Un fallecido |
| Chen et al.23 | China | 28 | 51 (24-73) | 64,2% | Sialoadenitis (79), dacrioadenitis (46) linfadenopatía (43), pancreatitis (32), colangitis (29) | 93% recibieron prednisona. 68% recibieron otros inmunosupresores. Eficacia del tratamiento 90%. Sin fallecimientos |
| Fernández-Codina et al.47 | España | 55 | 53 | 69,1% | Retroperitoneo (27), pseudotumor orbitario (22), glándulas salivales (16), páncreas (16%), glándulas lacrimales (16) | 85% recibieron corticosteroides y 34% otros inmunosupresores. 46% tuvieron respuesta completa y 50% parcial. Un fallecimiento por neumonía |
| Inoue et al.15 | Japón | 235 | 67 (35-86) | 80,4% | Páncreas (60), glándulas salivales (34), riñón (23), glándulas lacrimales (23), aorta (20) | 78% recibieron tratamiento con corticoides y todos ellos entraron en remisión. No se usaron otros inmunosupresores. 24% con recaídas |
| Wallace et al.16 | EE.UU. | 125 | 55 (24-83) | 60,8% | Submandibulares (28), ganglios linfáticos (27), órbita (22), páncreas (19), fibrosis retroperitoneal (18) | 51% recibieron corticoides, con repuesta en 86% de ellos pero recaída en 77%. 85% con enfermedad activa y 68% sin tratamiento al momento del registro |
El diagnóstico de la entidad se realiza generalmente entre la sexta y séptima década de la vida, afectando más frecuentemente al páncreas pero habiéndose descrito casos con compromiso de casi cualquier componente de la anatomía así como en población pediátrica11-14. Macroscópicamente y por imagen es posible documentar el aumento de tamaño del órgano afectado y la fibrosis del mismo, motivo por el cual previo a la era actual del diagnóstico inmunohistoquímico diversas manifestaciones de la misma enfermedad se asumieran como entidades diferentes con epónimos de acuerdo al órgano afectado. Hoy en día, tanto la tiroiditis de Riedel (tiroiditis fibrosante) como el tumor de Küttner (aumento de tamaño de glándulas submandibulares, con fibrosis), la enfermedad de Ormond (fibrosis retroperitoneal) y la de Mikulicz se clasifican dentro del espectro de ER-IgG4. La enfermedad de Mikulicz es el prototipo de afectación de órganos por esta entidad después del páncreas.
La ER-IgG4 se presenta predominantemente en hombres. Hasta un 39% de los pacientes tienen diagnóstico previo o concomitante de diabetes mellitus, probablemente por la afectación pancreática15. Además de la afectación pancreática, de las glándulas salivares y de las adenopatías, se describe frecuentemente afectación renal, aórtica, retroperitoneal y pulmonar. La frecuencia variable de estas manifestaciones en las cohortes pudiera deberse tanto a diferencias en predisposición genética o factores de riesgo desconocidos hasta la fecha, como a diferencias en los métodos empleados para el diagnóstico15,16. También existe cierta diferencia en las manifestaciones según el sexo del paciente (fig. 1). Una de las principales conclusiones del análisis de las cohortes es que el compromiso de un órgano aislado es la excepción, mas no la regla en ER-IgG4, tratándose en la mayor parte de los casos de una enfermedad multisistémica16.
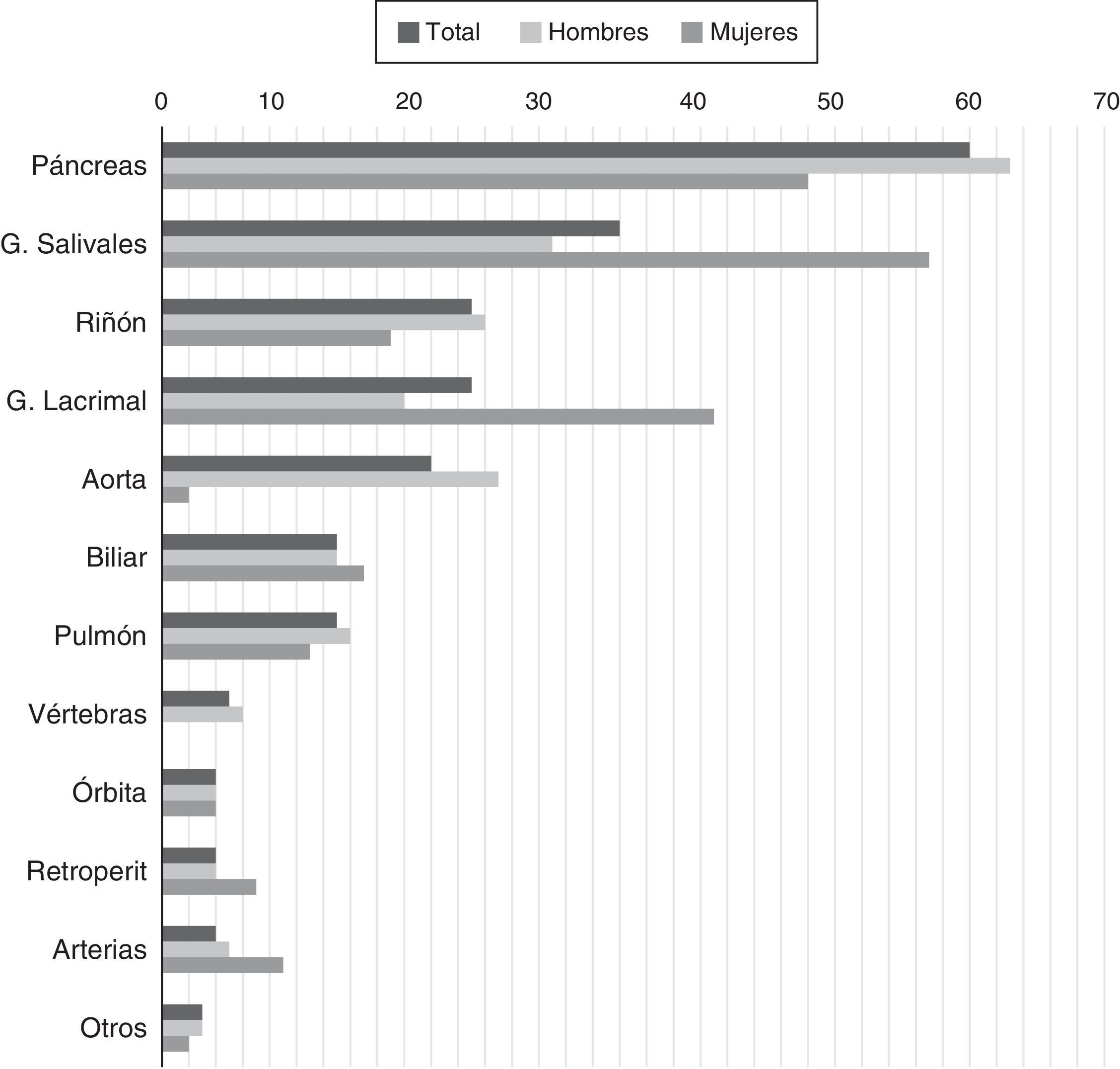
Porcentaje de pacientes con determinados órganos comprometidos debido a ER-IgG4 confirmada.
Fuente: adaptada de Hasosah et al.14.
A diferencia de otros tipos de pancreatitis, la ocasionada por ER-IgG4 no se relaciona con la presencia de autoanticuerpos circulantes. Se manifiesta generalmente con dolor abdominal, ictericia por edema e infiltración de los ductos pancreáticos y biliares, pérdida de peso e insuficiencia pancreática exocrina o endocrina17. El compromiso renal es más variado, describiéndose nefritis tubulointersticial, dolor por aumento de tamaño de los riñones, grados variables de hematuria y proteinuria o nefropatía obstructiva por fibrosis retroperitoneal18. En la afectación pulmonar se puede observar casi cualquier patrón radiológico, incluyendo: 1)afectación parenquimatosa localizada, 2)afectación parenquimatosa difusa, 3)presencia de adenopatías, 4)patrón intersticial difuso, 5)engrosamiento bronquial y 6)nódulos pulmonares. Dada la heterogeneidad de la afectación pulmonar, en ocasiones únicamente puede confirmarse mediante biopsia19. También son visibles por imagen los procesos fibróticos previamente mencionados en cuello, mediastino y abdomen20,21. Se ha descrito también la presencia simultánea o la aparición posterior de neoplasias en pacientes con diagnóstico de ER-IgG4, siendo en orden de frecuencia las neoplasias gástricas, las colorrectales y la neoplasia de próstata las más comunes de diagnóstico simultáneo y la neoplasia pulmonar las más frecuente de aparición posterior, aunque se desconoce a la fecha una relación causa/efecto entre ER-IgG4 y malignidad15.
Diagnóstico de la enfermedadLa presentación inicial de la enfermedad puede ser muy inespecífica o sugerir otras enfermedades mucho más comunes. Incluso en algunas ocasiones la IgG4 puede ir acompañada de otros procesos inflamatorios tales como enfermedades autoinmunes, vasculitis sistémicas o procesos neoplásicos, pero aún desconocemos las bases fisiopatogénicas para tal asociación.
En la cohorte francesa los pacientes consultaron predominantemente por síndrome constitucional, pérdida de peso y síntomas secos en un tercio de los casos. Al examen físico casi la mitad de ellos tenían linfadenopatías, pero solo una quinta parte tenían ictericia o aumento de tamaño de las glándulas salivales22. En la cohorte china, el aumento de tamaño de las glándulas salivales fue el síntoma o hallazgo predominante, seguido de historia de alergia en la mitad de los casos, dolor abdominal y linfadenopatías23.
Los estudios de imagen revelan aumento de tamaño del órgano afectado o compromiso fibrosante regional. La tomografía con emisión de positrones (PET), que es útil en lesiones hipermetabólicas (de naturaleza inflamatoria, no infecciosa o neoplásica), permite detectar el aumento del metabolismo en los órganos afectados pero no diferenciar entre las diversas etiologías con este patrón. Su utilidad estaría en documentar el compromiso multiorgánico24,25. Dentro de los hallazgos de laboratorio es frecuente encontrar niveles séricos elevados de IgG total (61%>1.800mg/dl), IgG4 (84%>135mg/dl) e IgE (58%>360mg/dl). La frecuencia de autoanticuerpos es similar a la esperada para el grupo etario en el que se presenta26.
Aunque la IgG4 otorga el nombre a la enfermedad, los niveles séricos de esta no se encuentran elevados en la totalidad de los pacientes en los cuales se llega a un diagnóstico por biopsia. Según los criterios de inclusión que se empleen en el estudio, la sensibilidad de la IgG4 sérica oscila entre un 50 y un 90%, con una especificidad altamente variable y tan baja como el 60%27,28. Una IgG4 elevada puede encontrarse en casos de diversas enfermedades respiratorias (bronquiectasias, asma, sinusitis crónica, sarcoidosis), biliares (colangitis esclerosante primaria, colangiocarcinoma, litiasis), pancreatitis crónica de otras etiologías y cirrosis. Entre las enfermedades autoinmunes, el síndrome de Sjögren, el lupus eritematoso sistémico, la artritis reumatoide, las miopatías inflamatorias y las vasculitis pueden también elevar el valor sérico de la inmunoglobulina26,29.
No existe un criterio internacionalmente aceptado para de diagnóstico de la ER-IgG4. Japón ha liderado la investigación y el conocimiento sobre la enfermedad, generando al menos 2 grupos de criterios diagnósticos (tabla 2). Por otro lado, posiblemente sea la suma de los criterios clínicos, histopatología y serología la que defina la probabilidad de que un paciente dado tenga la enfermedad, pudiendo darse por confirmada cuando se cumplan los 3 y descartada en su ausencia30 (fig. 2). El clínico debe orientarse a partir de las manifestaciones clínicas descritas, los hallazgos radiológicos de fibrosis, organomegalia o adenopatías, la elevación de niveles séricos de IgG4 en ausencia de otra explicación y los hallazgos patológicos característicos en tinciones usuales e inmunohistoquímica. Siempre debe buscarse el medio de realizar biopsia para excluir diagnósticos diferenciales31. Como se mencionó anteriormente, es mucho más probable encontrar una enfermedad multiorgánica que con compromiso de órgano aislado, por lo que siempre debe hacerse un estudio sistemático guiado por la clínica y alteraciones de laboratorio.
Dos grupos de criterio para el diagnóstico de ER-IgG4
| Criterios de Umehara et al.12 | Criterios de Okazaki et al.48,49 |
|---|---|
| Hallazgos clínicos altamente sugestivos Edema simétrico de glándulas lacrimales, parótidas o submandibulares, pancreatitis autoinmune, seudotumor inflamatorio, fibrosis retroperitoneal, sospecha de enfermedad de Castleman Hallazgos de laboratorio altamente sugestivos IgG4>135mg/dl Células IgG4+>40% del total de células IgG+ Hallazgos clínicos sugestivos Edema unilateral de glándula lacrimal, parótida o submandibular, pseudotumor orbital, colangitis esclerosante, prostatitis, paquimeningitis hipertrófica, neumonitis intersticial, nefritis intersticial, alteración tiroidea, hipofisitis, aneurisma inflamatorio Laboratorios sugestivos Hipergammaglobulinemia no explicada, hipocomplementemia, hiper IgE o eosinofilia, adenopatías por medicina nuclear | 1. Agrandamiento o lesiones focales o difusas en uno o más órganos 2. Concentraciones séricas de IgG4>135mg/dl 3. Histopatología a) Infiltrado linfocitario y plasmocítico con fibrosis, sin infiltrado neutrófilo b) Infiltrado de plasmocitos IgG4 positivos mayor de 10/cap o proporción de células IgG4/IgG>40% c) Fibrosis estoriforme-remolino d) Flebitis obliterativa Se realiza el diagnóstico con el cumplimiento de alguna de las siguientes combinaciones de criterios: • 1+2 • 1+3 (a+b) • 2+3 (a+b) • 3 (a+b+c+d) |
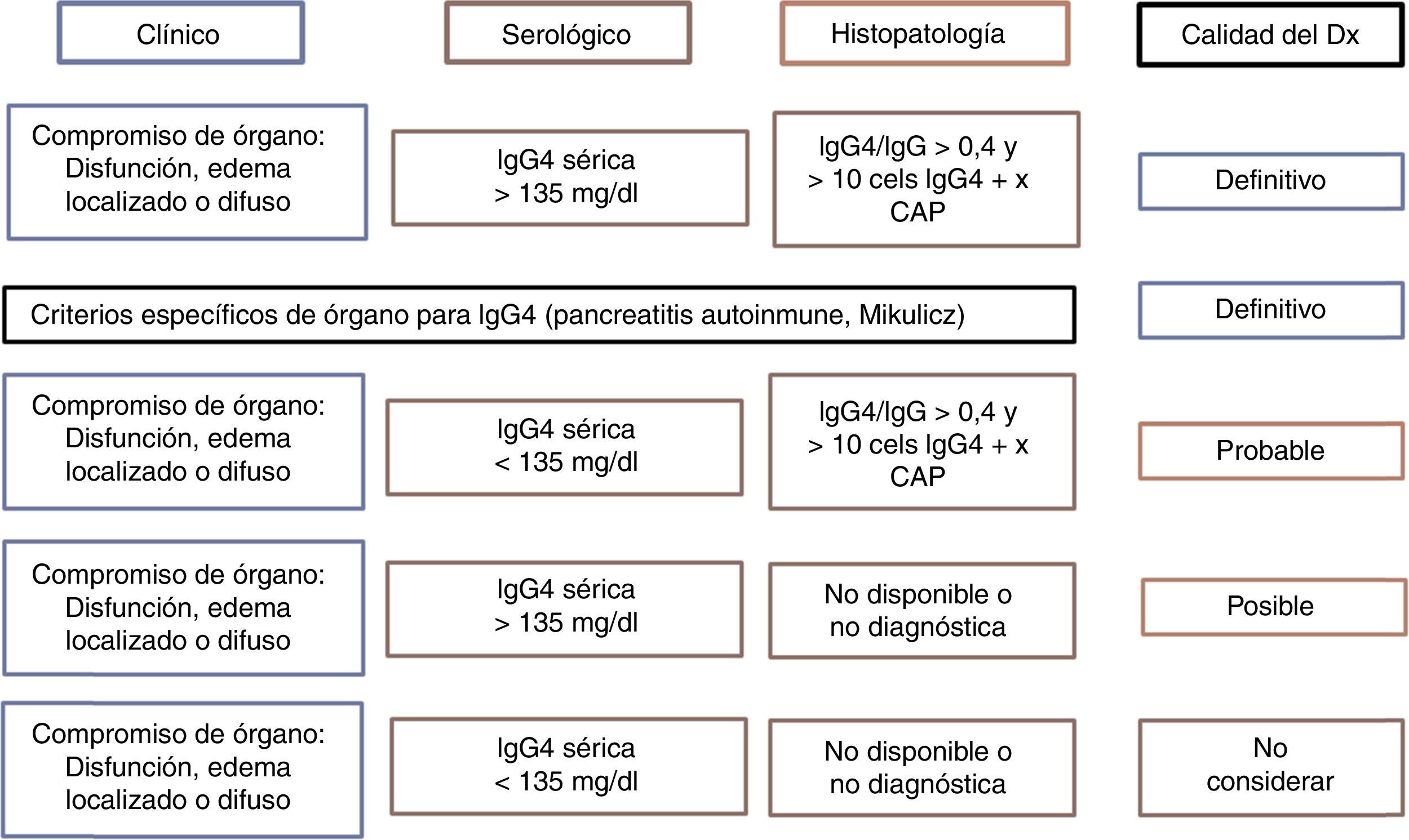
Certeza del diagnóstico de ER-IgG4 según criterios diagnósticos.
Fuente: adaptada de Ryu et al.29.
Dado que uno de los principales diagnósticos diferenciales de la enfermedad es el síndrome de Sjögren, el clínico debe tener en mente ciertos aspectos que permiten distinguir ambas enfermedades. A diferencia de la ER-IgG4, el síndrome de Sjögren es más común en mujeres, no presenta usualmente aumento de tamaño de las glándulas lacrimales, comienza con síntomas secos en gran parte de los casos, suele generar destrucción glandular por inflamación y tiene un perfil serológico de autoinmunidad claramente definido. En la histopatología, el criterio de clasificación del Sjögren exige un focus score de al menos1, definido como la presencia de al menos un foco mínimo de 50 linfocitos adyacentes a acinos glandulares de aspecto normal por cada 4mm2 de tejido, hallazgo muy diferente a lo mencionado previamente para ER-IgG432. Otros datos clínicos y de laboratorio útiles se consignan en la tabla 3. La lista completa de diagnósticos diferenciales es amplia e involucra diversas enfermedades con compromiso multisistémico, adenopatías generalizadas y hallazgos no específicos en ayudas diagnósticas. Los más comunes se incluyen en la tabla 4.
Diferencias clínicas y de laboratorio entre el síndrome de Sjögren y la ER-IgG4
| Hallazgo | Sjögren | ER IgG4 | p |
|---|---|---|---|
| Xeroftalmia (%) | 93 | 32 | < 0,001 |
| Xerostomía (%) | 87 | 37 | < 0,001 |
| Artralgia (%) | 48 | 15 | < 0,001 |
| Neumonitis (%) | 32 | 9,4 | NS |
| Nefritis (%) | 6 | 17 | NS |
| Rinitis alérgica (%) | 6 | 40 | < 0,001 |
| FR positivo (%) | 87 | 26 | < 0,001 |
| Anti Ro/La (%) | 99 | 1,6 | < 0,001 |
| IgE sérica (UI/dl) | 15 | 307 | < 0,001 |
| IgG4 sérica (UI/dl) | 23 | 697 | < 0,001 |
NS: estadísticamente no significativo.
Fuente: adaptado de Masaki et al.50.
Principales diagnósticos diferenciales de la ER-IgG4
| Enfermedades reumatológicas | Enfermedades no reumatológicas | Malignidades y otras |
|---|---|---|
| Síndrome de Sjögren | Colangitis esclerosante | Linfomas (incluyendo tipo MALT) |
| Lupus eritematoso sistémico | Orbitopatía tiroidea | Enfermedad de Kikuchi-Fujimoto |
| Granulomatosis con poliangeítis | Tuberculosis | Enfermedad de Kimura |
| Arteritis de grandes vasos | Micosis invasivas | Enfermedad de Castleman |
| Sarcoidosis | Pancreatitis crónica de otras etiologías | Malignidades sólidas (en caso de manifestarse como masas aisladas o con apariencia de metástasis) |
| Enfermedad pulmonar intersticial primaria o secundaria |
La IgG4 constituye en condiciones normales menos del 5% de la IgG total, siendo el subtipo más infrecuente, pero elevando su cantidad relativa hasta el 80% en alergias crónicas18. Su estructura consiste, al igual que el resto de subtipos, en 2 cadenas pesadas y 2 ligeras, pero los puentes disulfuro que unen las 2 cadenas pesadas son inestables, permitiendo su separación y mezcla con otros fragmentos de IgG4, generando así moléculas divalentes33. Esta inmunoglobulina tiene una pobre capacidad para activar el complemento y formar complejos inmunes, por lo que se ha postulado que tiene un papel antiinflamatorio al bloquear competitivamente otros subtipos mucho más activos34,35. A pesar de esto, se ha visto implicada en otras 3 enfermedades: pénfigo foliáceo (en la cual el subtipo predomina en los depósitos subepidérmicos), púrpura trombocitopénica trombótica (en la que aumentan sus niveles circulantes) y algunos casos de glomerulonefritis membranosa1. Su papel como causante de la fibrosis en ER-IgG4 es mucho menos claro. Un modelo de patogénesis de la enfermedad señala que algún desencadenante desconocido (infección viral o antígeno no identificado) desencadena una respuesta inmunológica provocando una infiltración del órgano por linfocitosB que se diferencian en células plasmáticas. En el caso específico del páncreas la selección de las células IgG4+ estaría a cargo de los miofibroblastos (células estrelladas). Este modelo explica la infiltración, pero no la fibrosis del tejido36. La infiltración del tejido no solo es por células plasmáticas y linfocitosB, sino que se encuentran gran cantidad de linfocitos CD4. Estas células y sus mediadores (interleucinas, TNF) podrían estimular la proliferación del tejido fibroso, pero el causante del estímulo a los CD4 tampoco queda esclarecido. La anhidrasa carbónica tipoii y iv o algunas enzimas pancreáticas podrían servir como antígenos para la infiltración del órgano por células inflamatorias, pero estos no se encuentran en todos los tejidos que se pueden afectar por la enfermedad37. El perfil de citoquinas circulantes es usualmente Th2, explicando así el aumento de IgG4 sérica y depositada, pero la cantidad de células Th2 no se ve afectada por el tratamiento con corticosteroides, independientemente de la respuesta clínica38. A pesar del conocimiento adquirido, quedan aún preguntas por resolver: ¿cuál es el autoantígeno, si lo hay, desencadenante de la enfermedad? ¿Cuál es el mecanismo exacto de la fibrosis del tejido? ¿Tiene algún papel la IgG4 depositada?
Tratamiento de la enfermedad relacionada con IgG4 y seguimiento de los pacientesExiste un consenso internacional reciente sobre el tratamiento de la enfermedad (sin la existencia de uno sobre el diagnóstico). El clínico debe preguntarse en primera instancia si el paciente requiere tratamiento, con una respuesta afirmativa en la gran mayoría de casos. Los argumentos a favor incluyen la mejor respuesta y pronóstico del órgano afectado con el inicio temprano de la terapia, la posibilidad de tener compromiso multiorgánico que pudiera beneficiarse también de tratamiento y la respuesta generalmente favorable que existe con el mismo. Las excepciones incluirían pacientes con enfermedad mínimamente sintomática o sin mayores implicaciones pronósticas (aumento aislado y no limitante de tamaño de glándulas salivares o lacrimales, adenopatías), en los cuales se excluye compromiso de otros órganos, además de pacientes con compromiso predominantemente fibroso, que se benefician más de la intervención quirúrgica que de la farmacológica (seudotumores orbitales, mesenteritis esclerosante)31. Debe tenerse en cuenta que la enfermedad tiene un carácter impredecible en los órganos que comprometerá, y que pacientes con remisión inducida o espontánea pueden tener recaídas en otros sistemas39. Además, un retraso en el tratamiento por cualquier causa puede generar secuelas graves e irreversibles40.
La enfermedad responde usualmente a dosis altas de glucocorticoides, empleándose en promedio 40mg/día y realizando una disminución gradual de la misma31. La definición de «respuesta» al tratamiento difiere según la bibliografía, e incluso existe una escala diseñada para evaluarla, pero en la práctica se conceptúa a criterio del médico tratante41. La recaída de la enfermedad es bastante frecuente. En una cohorte de 116 pacientes con pancreatitis autoinmune tipoi (una manifestación de IgG4), la dosis inicial de prednisolona fue de 40mg, con un descenso gradual de 5mg semanales hasta la suspensión. La mitad de los pacientes tuvo recaía de la enfermedad42. El consenso internacional recomienda una disminución mucho más gradual de la dosis, en un periodo de 3 a 6 meses. Con este esquema, las recaídas fueron del 23% en la cohorte estadounidense16. Los japoneses mantuvieron en su cohorte los corticoides a dosis bajas (2,5-5mg/día) sin uso de ahorrador de corticoides hasta durante 3años, con recaídas reportadas del 24%15. El tratamiento propuesto para las recaídas iniciales es un nuevo ciclo de corticoides similar al inicial.
Respecto al uso de ahorradores de corticoides, existen 3 alternativas: emplearlos desde el inicio junto con los corticoides, iniciarlos al presentarse la primera recaída o no emplearlos en absoluto. En la cohorte anteriormente mencionada de tratamiento de pancreatitis autoinmune se empleó azatioprina, 6-mercaptopurina o micofenolato mofetilo luego de la primera recaída, sin que se observaran diferencias en el tiempo hasta una segunda recaída respecto a quienes recibieron únicamente un nuevo ciclo de corticoides (70% vs 60% a 48meses)42. Para los 3 medicamentos, la suspensión por efectos adversos se dio en el 17, el 33 y el 0% de los pacientes, respectivamente. En la cohorte estadounidense solo un paciente recibió ahorradores de corticoides, mientras que ninguno lo hizo en la japonesa, con los resultados anteriormente mencionados. En el consenso internacional, solo un 46% de los expertos consultados estuvo de acuerdo en el uso de uno de estos 3 medicamentos en algún momento del tratamiento, sin recomendaciones específicas respecto a su indicación.
Recientemente se publicaron resultados muy favorables con el uso de rituximab (RTX) como terapia de inducción y/o mantenimiento. Un estudio reciente evaluó la eficacia del RTX en 12 pacientes a dosis de 375mg/m2 semanal durante 4 semanas, seguido de nuevas dosis de acuerdo al criterio del médico tratante. La indicación para su uso fue la suspensión por fallo terapéutico de los ahorradores o intolerancia a los corticoides. Se definió remisión completa si el paciente presentaba resolución clínica, radiográfica y bioquímica a nivel pancreático o en el órgano afectado, ausencia de nuevas lesiones de inflamación durante el seguimiento y suspensión de las terapias de mantenimiento para el control de la enfermedad. Se definió como remisión parcial si el paciente presentaba mejoría pero sin resolución de la inflamación (clínica, radiográfica o bioquímica) sin la necesidad de corticoides, y remisión incompleta cuando se alcanzaba una mejoría de los cambios inflamatorios pero con la necesidad de tratamiento concomitante con RTX y corticoides. Diez (83%) de los 12 pacientes alcanzaron una remisión completa, un paciente alcanzó una remisión parcial y un paciente, una remisión incompleta42.
Otro estudio empleó el RTX como terapia de inducción a dosis de 1.000mg en los días 1 y 15, sin corticoides simultáneos (26 pacientes) o con descenso gradual de estos en los primeros 2meses (4pacientes). Empleando la escala de actividad de ER-IgG4, un 97% de los individuos obtuvieron respuesta de la actividad a los 6 meses, con un 47% en respuesta completa y un 40% manteniéndola a los 12meses43. La dosis inicial de RTX parece ser de gran importancia, dado que en otro estudio una dosis única de 500mg de RTX con un esquema de descenso progresivo de la dosis de corticoides se relacionó con un número elevado de recaídas y ausencia de respuesta44. Se ha empleado también el metotrexato como terapia de sostenimiento junto con corticoides, con resultados aceptables (50% de remisión de enfermedad a los 24meses), pero sin un grupo de comparación u otro estudio que permita obtener conclusiones sobre el uso de este fármaco en ER-IgG445.
El seguimiento de los pacientes es eminentemente clínico, con apoyo de técnicas de laboratorio usuales según el órgano afectado, además de métodos de imagen31. Se ha propuesto además el seguimiento de niveles séricos de IgG4, que disminuye con el inicio del tratamiento y cuyo aumento posterior parece correlacionarse con recaídas44,46. Experimentalmente se ha empleado además la medición de la cantidad de plasmoblastos circulantes y niveles séricos de CD19, pero su uso y su utilidad en la práctica clínica aún no están estandarizados38.
ConclusionesLa ER-IgG4 es una entidad recientemente descrita, de etiología y fisiopatología aún no aclaradas, caracterizada por compromiso fibrótico e infiltrado linfoplasmocitario con predominio de células IgG4+ en uno o varios órganos blanco, siendo los más comúnmente afectados el páncreas, los ganglios linfáticos y las glándulas salivales, pero con capacidad para afectar la mayor parte de estructuras de la anatomía, cursando en la mayoría de casos con niveles séricos elevados de IgG4. Pese a su nombre, no se ha definido claramente el papel que juega la IgG4 en la enfermedad, el estímulo que genera infiltración de órgano por células inflamatorias ni los factores que determinan cuál o cuáles órganos se verán involucrados en la entidad. Ante la ausencia de un criterio diagnóstico fehaciente, es la suma de argumentos la que permite el diagnóstico (presentación clínica, apariencia en imágenes, IgG4 sérica elevada, histopatología, respuesta a corticoides). Su tratamiento actual consiste en el uso de dosis altas de corticoides sistémicos, con regímenes de descenso progresivo de dosis. No existe evidencia que sustente el uso de medicación ahorradora de corticoides en la actualidad, pero la terapia biológica con RTX se ha empleado con éxito en la inducción y el sostenimiento de respuesta clínica, por lo que se plantea como terapia de segunda línea.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
FinanciaciónJose A. Gómez-Puerta recibe apoyo de Colciencias (convocatoria 656 de 2014).
AutoríaOAS y JAGP participaron en la concepción, diseño, análisis e interpretación de los datos, redacción, revisión y aprobación del manuscrito remitido. AA participó en el análisis e interpretación de los datos, redacción, revisión y aprobación del manuscrito remitido.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


