





Las miopatías inflamatorias idiopáticas son un grupo heterogéneo de enfermedades autoinmunitarias adquiridas del músculo estriado que se caracterizan por debilidad muscular, elevación de enzimas musculares, anormalidades electromiográficas e identificación de infiltrado inflamatorio en la biopsia muscular. En este grupo se incluyen la polimiositis, la dermatomiositis y la miositis por cuerpos de inclusión. Se consideran dentro del grupo de enfermedades autoinmunitarias poco frecuentes debido a su baja incidencia global de 2–10 casos por millón de habitantes por año, y presentan diferentes patrones de presentación por edad, sexo y raza. La etiología se considera desconocida, siendo los factores genéticos, aunados a la exposición de algunos agentes ambientales, los que pudieran desencadenar una respuesta autoinmunitaria teniendo como órgano diana el músculo esquelético.
Idiopathic inflammatory myopathies are a group of heterogeneous striated muscle acquired autoimmune diseases, characterized by progressive symmetrical muscle weakness, elevated serum levels of muscle enzymes, electromyographic abnormalities and inflammatory infiltrates on muscle biopsy. This group of diseases comprises polymyositis, dermatomyositis and inclusion-body myositis. They are considered rare autoimmune diseases, with an overall incidence range of 2 to10 new cases per million persons at risk per year, with differences in distribution according to age, gender and race. Their etiology is largely unknown, but it likely involves both genetic and environmental factors that contribute to autoimmune disorders, with striated muscle as a common target.
Las miopatías inflamatorias idiopáticas (MII) son un grupo heterogéneo de enfermedades autoinmunitarias adquiridas del músculo estriado, en el que se incluyen la polimiositis (PM), dermatomiositis (DM) y miositis por cuerpos de inclusión (MCI)1. Cursan de forma aislada o asociadas a otras enfermedades autoinmunitarias sistémicas o secundarias a neoplasias. Tienen un curso clínico variable con una manifestación clínica común: debilidad muscular proximal, en general con elevación de enzimas musculares, patrón miopático en la electromiografía1,2 y biopsia muscular característica.
EpidemiologíaLa incidencia global de las miopatías inflamatorias se reporta en rangos de 2–10 casos por millón de habitantes por año2,3. La mayoría de los estudios realizados son en poblaciones anglosajonas y utilizan los criterios de clasificación de Bohan y Peter. Se han encontrado reportes de hasta 4,9 a 8,4 casos/millón asociados a mayor sospecha clínica y mejoría de técnicas diagnósticas más que a un incremento real de la incidencia misma2,3. La prevalencia se estima en 8/100.000 habitantes4.
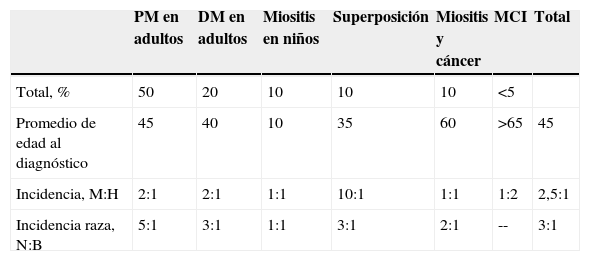
Se sabe que el patrón de incidencia cambia de acuerdo con los grupos de edad, sexo y grupo étnico (tabla 1). Los rangos de incidencia anual se incrementan con la edad: de 2,5 por millón en menores de 15 años a 10,5 por millón en mayores de 65 años4. Sin embargo, se han establecido picos de incidencia bimodal, siendo en niños de 5 y 14 años predominante la DM, y en adultos de 45 y 64 años generalmente la PM. En la MCI se reporta una mayor prevalencia en pacientes mayores de 50 años5.
Características epidemiológicas de las miopatías inflamatorias
| PM en adultos | DM en adultos | Miositis en niños | Superposición | Miositis y cáncer | MCI | Total | |
| Total, % | 50 | 20 | 10 | 10 | 10 | <5 | |
| Promedio de edad al diagnóstico | 45 | 40 | 10 | 35 | 60 | >65 | 45 |
| Incidencia, M:H | 2:1 | 2:1 | 1:1 | 10:1 | 1:1 | 1:2 | 2,5:1 |
| Incidencia raza, N:B | 5:1 | 3:1 | 1:1 | 3:1 | 2:1 | -- | 3:1 |
Tomado de Oddis C, Medsger T. Clinical features, classification and epidemiology of inflammatory muscle disease. En: Hochberg M, Silman A, Smolen J, editores. Rheumatology. 4th ed. Mosby. 2008;10:1433–8.
B: raza blanca; DM: dermatomiositis; H: hombre; M: mujer; MCI: miositis por cuerpos de inclusión; N: raza negra; PM: polimiositis.
El rango de incidencia por sexo (mujer:hombre) es de 2,5:1, siendo cercana a 1:1 en la niñez y en los casos de asociación a neoplasia, y tan alto como 10:1 cuando coexiste con otra enfermedad del tejido conectivo. En la MCI la relación se invierte, siendo de 1:22,3. Dentro de las diferencias raciales, se han encontrado formas de inicio adulto más tempranas en afroamericanos en comparación con población anglosajona, presentando mayor incidencia de PM además de predominio femenino (17,1 por millón vs. 6,1 por millón, respectivamente)2. Shamim et al6 publicaron un artículo con referencia al grupo de mestizos mesoamericanos, que mostraba una mayor frecuencia de DM en este tipo de población en comparación con la caucásica, coincidiendo con la mayor cercanía a la línea del ecuador así como con una mayor exposición a la radiación ultravioleta.
EtiologíaLa causa actualmente es desconocida. Existen algunas teorías sobre la interacción de agentes externos físicos, químicos y/o infecciosos que actúan en un territorio genético predispuesto, resultando en alteraciones autoinmunitarias7. Un estudio realizado por Okada et al8, que incluyó a 919 pacientes con DM o PM, demostró que la proximidad a la latitud 0° era un factor de riesgo para DM sobre la base de que los países más cercanos a la línea del ecuador presentan mayor exposición a radiación ultravioleta, siendo éste un factor de riesgo para desarrollar esta patología. Otros estudios relacionan los picos estacionales de marzo-abril con el desarrollo de miopatía, principalmente asociado a síndrome antisintetasa9. Sin embargo, existe también mayor número de casos principalmente en niños en el invierno, lo cual se ha atribuido a algún factor infeccioso viral o bacteriano10. Otros estudios han encontrado historia de infecciones respiratorias y gastrointestinales tratadas con antibióticos, previo al inicio de la DM juvenil11. Hasta el momento no se ha logrado aislar un agente infeccioso viral, aunque se han encontrado asociaciones con PM y MCI principalmente con retrovirus (VIH y HTLV-1 [virus linfotrópico de células T humano-1])12.
Dentro de los factores genéticos existe una fuerte asociación de los genes de HLA DRB1*0301 con todas las formas de MII13. Sin embargo, se ha asociado la presencia de HLA DRB1*0501 con DM juvenil, los polimorfismos del factor de necrosis tumoral 308A y del antagonista del receptor de interleucina 1 con la fotosensibilidad observada en la DM y el haplotipo HLA B8/DR3/DR52/DQ2 asociado en el 65% de los pacientes con MCI14.
Se han implicado algunos factores genéticos en el desarrollo etiológico de la autoinmunidad. Recientemente se ha encontrado elevación del interferón α y β en los estudios de inmunohistoquímica en biopsias de pacientes con DM, atribuyéndolo como factor etiológico de lesión tisular directa15.
Varios fármacos se han asociado con el desarrollo de miopatías inflamatorias. Dentro de los más importantes para considerar están D-penicilamina, cloroquina, ciclosporina, colchicina, esteroides e inhibidores de 3-hidroxi-metilglutaril coenzima A, en especial en combinación con fibratos o ácido nicotínico. Sin embargo, debe considerarse que existen algunos mecanismos idiosincráticos para el desarrollo de ésta que aún no se han dilucidado12,16.
La evidencia de autoinmunidad es frecuente con miositis debido a la presencia de títulos altos de autoanticuerpos observados a nivel sérico de entre el 60–80%, sin embargo, se han asociado principalmente con el desarrollo de subtipos clínicos y el pronóstico más que como agentes etiológicos17.
Diversos estudios epidemiológicos establecen sin lugar a dudas la asociación entre miopatías y neoplasias7 (excepto con MCI)18, considerándose en algunas ocasiones como factor predisponente fisiopatológico para el desarrollo de autoinmunidad al alterar la autotolerancia como efecto paraneoplásico17. Por tanto, debe considerarse la búsqueda intencionada de neoplasias ante la sospecha diagnóstica18.
ClasificaciónAunque se ha propuesto un gran número de clasificaciones para las MII conforme avanza el conocimiento de éstas, durante mucho tiempo prevaleció la propuesta por Bohan y Peter, descrita en 1975, que describe 5 grupos de miopatías (tabla 2), considerada actualmente obsoleta, ya que no incluye a las MCI ni considera una perspectiva clinicopatológica. Al respecto, Urbano et al proponen en 1991 una clasificación que integra propuestas anteriores de Karpati (1988)19 y Dalakas (1991)20, incluyendo la MCI y algunos subtipos desde una perspectiva clinicopatológica2 (tabla 3).
Clasificación de miopatías (Bohan y Peter)
| Grupo I | Polimiositis primaria idiopática |
| Grupo II | Dermatomiositis primaria idiopática |
| Grupo III | Miopatía asociada a neoplasia |
| Grupo IV | Miopatía de la infancia |
| Grupo V | Miopatía asociada a enfermedad del tejido conectivo |
Tomado de N Eng J Med. 1975;292:344–7.
Clasificación clinicopatológica de las miopatías inflamatorias
|
Tomado de Mastaglia F, Philips B. Idiopathic inflamatory myopathies: Epidemiology, classification and diagnostic criteria. Rheum Dis Clin of North Am. 2002;28:723–41.
En 1994, Miller propone una clasificación serológica de las miopatías inflamatorias, donde se incluyen los autoanticuerpos específicos de miositis y los autoanticuerpos asociados a miositis, y en la que además se sugieren presentaciones clínicas y pronósticas asociadas a la presencia de determinados autoanticuerpos21 (tabla 4).
Clasificación serológica de las miopatías inflamatorias*
| Categoría serológica | Asociaciones y comentarios |
| Autoanticuerpos específicos de miositis | |
| Antisintetasa (anti-Jo-1) | Artritis simétrica no erosiva, enfermedad pulmonar intersticial, fiebre, manos de mecánico, fenómeno de Raynaud, asociado a HLA-DR3, DQA1*0501. Respuesta moderada al tratamiento |
| Anti-SRP | Asociado a trastorno cardíaco, palpitaciones y mialgias. HLA-DR5, DQA1*0301, inicio muy agudo de polimiositis intensa. Respuesta deficiente al tratamiento |
| Anti-Mi-2 | Dermatomiositis clásica con signo V, signo del chal y proliferación cuticular. HLA DR7, DQA1*0201. Buena respuesta al tratamiento |
| Autoanticuerpos asociados a miositis | |
| Anti-PM-Scl | Alta incidencia de síndrome de superposición, esclerodermia y miositis. HLA DR3, DQw2 |
| Anti-Ku | Se observa en síndromes de superposición, esclerodermia y miositis. |
| Anti-U1RNP | Se observa en la miositis de los síndromes de superposición. |
| Anti-U2RNP | Asociado a síndromes de superposición, esclerodermia y miositis |
| Anti-Ku | Se observa en síndromes de superposición, esclerodermia y miositis. |
| Anti-U1RNP | Se observa en la miositis de los síndromes de superposición. |
| Anti-U2RNP | Asociado a síndromes de superposición, esclerodermia y miositis |
SRP: signal recognition particle, partícula de reconocimiento de señal.
En 1996, Withmore et al describen una clasificación con el fin de incluir el subgrupo de DM sin miositis, también denominada DM amiopática, descrita por Krain en 197522, que incluye a los pacientes con lesiones cutáneas características y sin afección muscular durante al menos 2 años de seguimiento. Y por último, se describe la última clasificación de las enfermedades musculares inflamatorias, descrita por Mastaglia, donde se separan con claridad las formas infecciosas de las autoinmunitarias generalizadas y focales18 (tabla 5).
Clasificación de las miopatías inflamatorias
| A. Formas infecciosas |
| • Virales (coxsackie B, influenza A y B, VIH) |
| • Bacterianas (Streptococcus, Staphylococcus, Clostridium, TB) |
| • Micóticas (candidiasis, coccidioidomicosis) |
| • Protozoarios (toxoplasmosis, sarcocistosis) |
| • Helmintos (triquinosis, cisticercosis, haycocknemia) |
| B. Formas autoinmunitarias (idiopáticas) |
| Generalizadas |
| • Miositis por cuerpos de inclusión |
| • Dermatomiositis |
| • Polimiositis |
| • Síndromes de superposición |
| • Miopatía necrotizante (paraneoplásica) |
| • Miositis eosinofílica |
| • Miositis granulomatosa |
| Formas focales |
| • Miositis nodular localizada |
| • Miositis monomélica |
| • Miositis angiopática |
| • Miositis eosinofílica |
| • Miofascitis macrofágica |
| • Pseudotumor inflamatorio |
| • Miositis orbitaria |
Tomado de Mastaglia FL. Inflammatory muscle diseases. Neurol India 2008;56:263–70.
TB: tuberculosis; VIH: virus de la inmunodeficiencia humana.


